Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMessage-passing neural networks for high-throughput polymer screening
Jul 26, 2018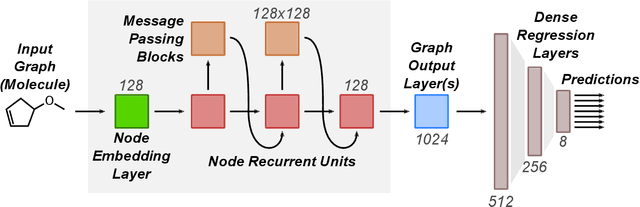
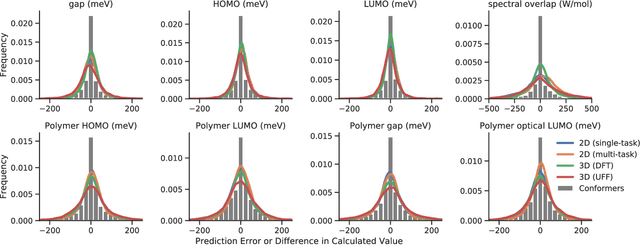
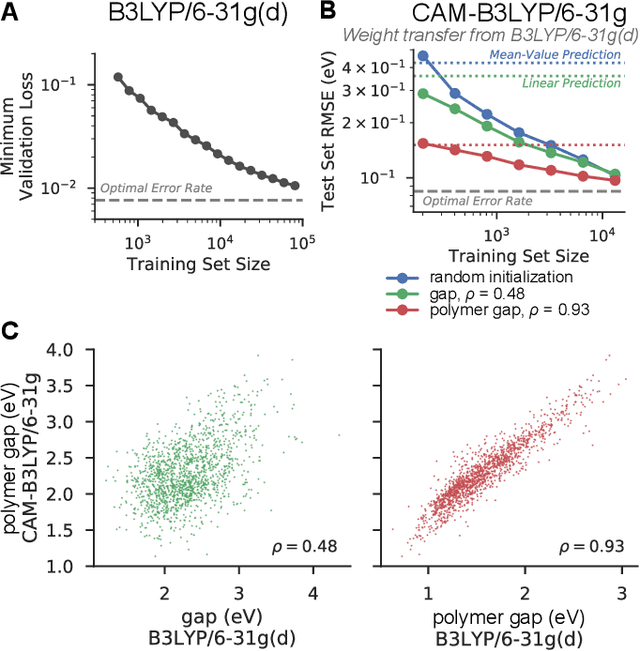
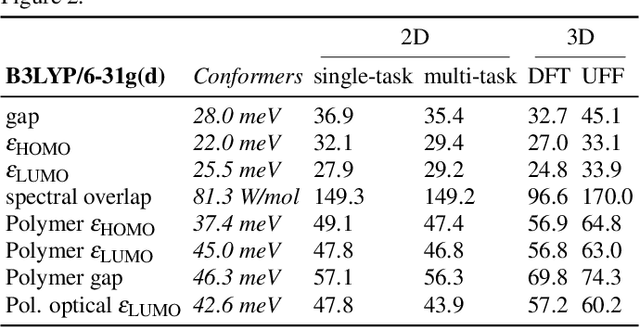
Machine learning methods have shown promise in predicting molecular properties, and given sufficient training data ML may surpass density functional theory in computational speed and chemical accuracy. However, the most accurate machine learning methods require optimized 3D molecular geometries, limiting their applicability for high-throughput screening. We show that near-optimal results for large polymeric molecules can be obtained without optimized 3D geometry, and that trained model weights can be used to improve performance on related tasks.
* 10 pages, 3 figures
Via