 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStochastic-based Neural Network hardware acceleration for an efficient ligand-based virtual screening
Jun 03, 2020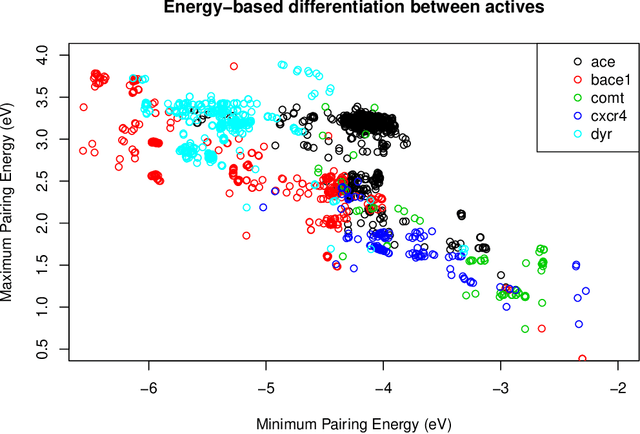
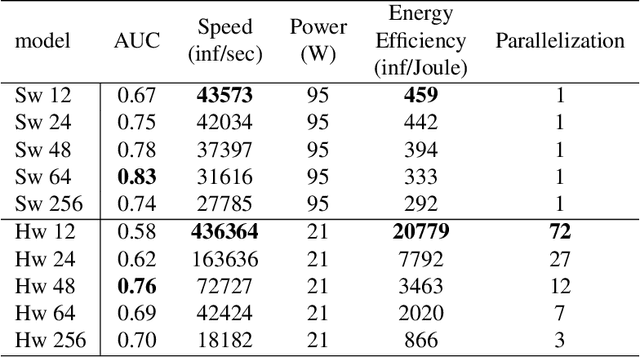
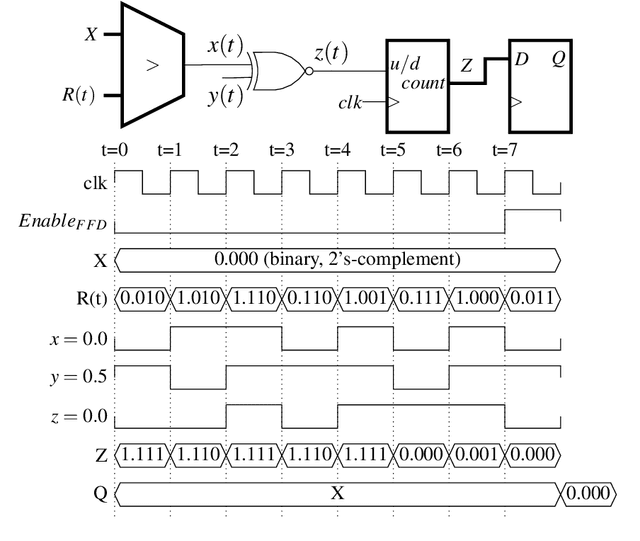
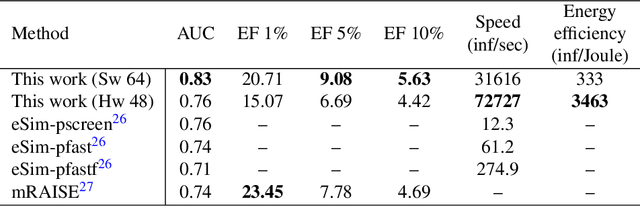
Artificial Neural Networks (ANN) have been popularized in many science and technological areas due to their capacity to solve many complex pattern matching problems. That is the case of Virtual Screening, a research area that studies how to identify those molecular compounds with the highest probability to present biological activity for a therapeutic target. Due to the vast number of small organic compounds and the thousands of targets for which such large-scale screening can potentially be carried out, there has been an increasing interest in the research community to increase both, processing speed and energy efficiency in the screening of molecular databases. In this work, we present a classification model describing each molecule with a single energy-based vector and propose a machine-learning system based on the use of ANNs. Different ANNs are studied with respect to their suitability to identify biochemical similarities. Also, a high-performance and energy-efficient hardware acceleration platform based on the use of stochastic computing is proposed for the ANN implementation. This platform is of utility when screening vast libraries of compounds. As a result, the proposed model showed appreciable improvements with respect previously published works in terms of the main relevant characteristics (accuracy, speed and energy-efficiency).
Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties
Mar 18, 2013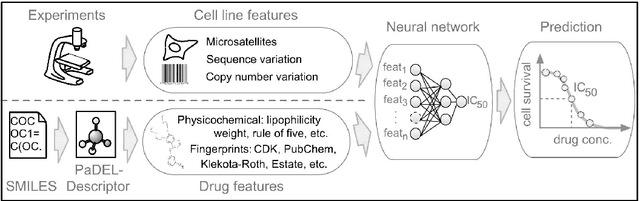
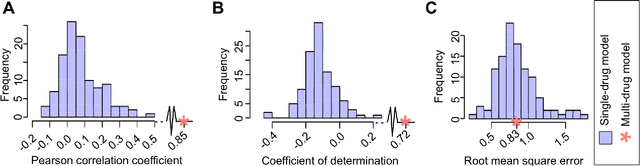
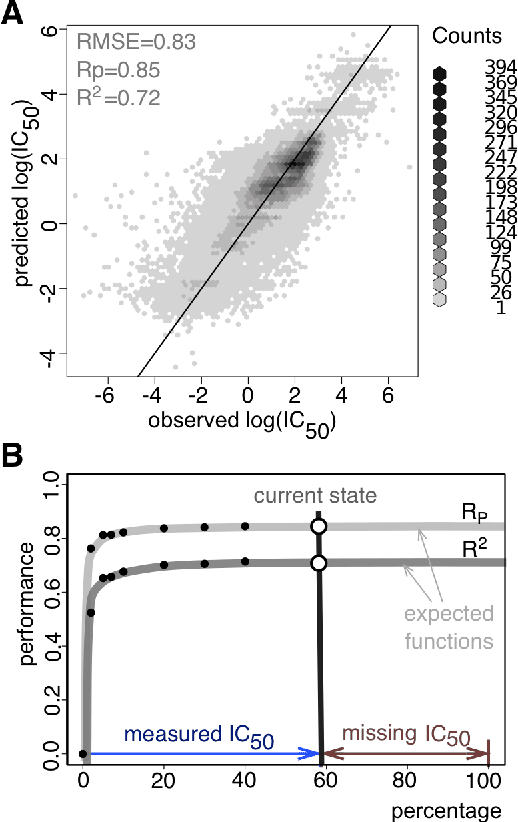
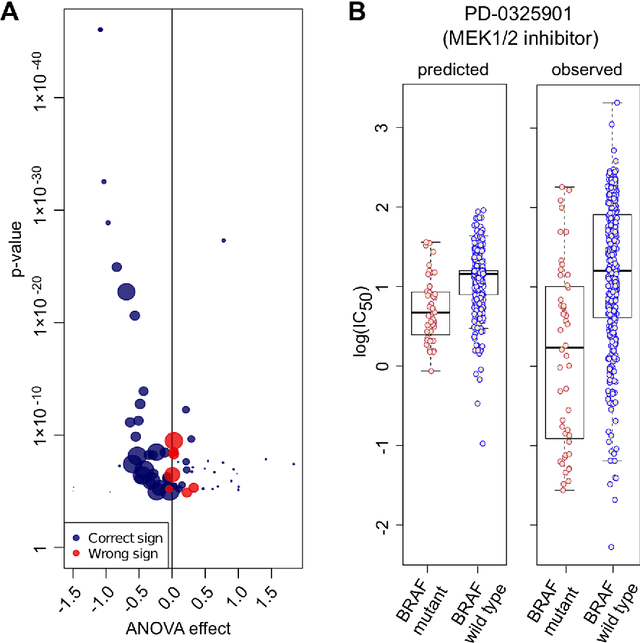
Predicting the response of a specific cancer to a therapy is a major goal in modern oncology that should ultimately lead to a personalised treatment. High-throughput screenings of potentially active compounds against a panel of genomically heterogeneous cancer cell lines have unveiled multiple relationships between genomic alterations and drug responses. Various computational approaches have been proposed to predict sensitivity based on genomic features, while others have used the chemical properties of the drugs to ascertain their effect. In an effort to integrate these complementary approaches, we developed machine learning models to predict the response of cancer cell lines to drug treatment, quantified through IC50 values, based on both the genomic features of the cell lines and the chemical properties of the considered drugs. Models predicted IC50 values in a 8-fold cross-validation and an independent blind test with coefficient of determination R2 of 0.72 and 0.64 respectively. Furthermore, models were able to predict with comparable accuracy (R2 of 0.61) IC50s of cell lines from a tissue not used in the training stage. Our in silico models can be used to optimise the experimental design of drug-cell screenings by estimating a large proportion of missing IC50 values rather than experimentally measure them. The implications of our results go beyond virtual drug screening design: potentially thousands of drugs could be probed in silico to systematically test their potential efficacy as anti-tumour agents based on their structure, thus providing a computational framework to identify new drug repositioning opportunities as well as ultimately be useful for personalized medicine by linking the genomic traits of patients to drug sensitivity.