 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Metric for the Balance of Information in Graph Learning
Jan 31, 2025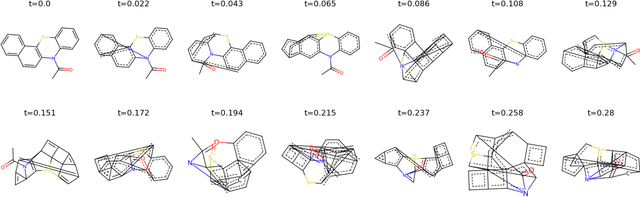
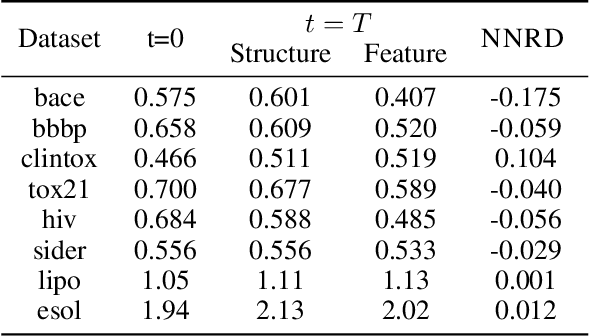

Graph learning on molecules makes use of information from both the molecular structure and the features attached to that structure. Much work has been conducted on biasing either towards structure or features, with the aim that bias bolsters performance. Identifying which information source a dataset favours, and therefore how to approach learning that dataset, is an open issue. Here we propose Noise-Noise Ratio Difference (NNRD), a quantitative metric for whether there is more useful information in structure or features. By employing iterative noising on features and structure independently, leaving the other intact, NNRD measures the degradation of information in each. We employ NNRD over a range of molecular tasks, and show that it corresponds well to a loss of information, with intuitive results that are more expressive than simple performance aggregates. Our future work will focus on expanding data domains, tasks and types, as well as refining our choice of baseline model.
Its All Graph To Me: Foundational Topology Models with Contrastive Learning on Multiple Domains
Nov 07, 2023



Representations and embeddings of graph data have been essential in many domains of research. The principle benefit of learning such representations is that the pre-trained model can be fine-tuned on smaller datasets where data or labels are scarse. Existing models, however, are domain specific; for example a model trained on molecular graphs is fine-tuned on other molecular graphs. This means that in many application cases the choice of pre-trained model can be arbitrary, and novel domains may lack an appropriate pre-trained model. This is of particular issue where data is scarse, precluding traditional supervised methods. In this work we use adversarial contrastive learning to present a \method, a model pre-trained on many graph domains. We train the model only on topologies but include node labels in evaluation. We evaluate the efficacy of its learnt representations on various downstream tasks. Against baseline models pre-trained on single domains, as well as un-trained models and non-transferred models, we show that performance is equal or better using our single model. This includes when node labels are used in evaluation, where performance is consistently superior to single-domain or non-pre-trained models.
Hierarchical GNNs for Large Graph Generation
Jun 20, 2023



Large graphs are present in a variety of domains, including social networks, civil infrastructure, and the physical sciences to name a few. Graph generation is similarly widespread, with applications in drug discovery, network analysis and synthetic datasets among others. While GNN (Graph Neural Network) models have been applied in these domains their high in-memory costs restrict them to small graphs. Conversely less costly rule-based methods struggle to reproduce complex structures. We propose HIGGS (Hierarchical Generation of Graphs) as a model-agnostic framework of producing large graphs with realistic local structures. HIGGS uses GNN models with conditional generation capabilities to sample graphs in hierarchies of resolution. As a result HIGGS has the capacity to extend the scale of generated graphs from a given GNN model by quadratic order. As a demonstration we implement HIGGS using DiGress, a recent graph-diffusion model, including a novel edge-predictive-diffusion variant edge-DiGress. We use this implementation to generate categorically attributed graphs with tens of thousands of nodes. These HIGGS generated graphs are far larger than any previously produced using GNNs. Despite this jump in scale we demonstrate that the graphs produced by HIGGS are, on the local scale, more realistic than those from the rule-based model BTER.