 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDifferentiable Rotamer Sampling with Molecular Force Fields
Feb 22, 2023Molecular dynamics is the primary computational method by which modern structural biology explores macromolecule structure and function. Boltzmann generators have been proposed as an alternative to molecular dynamics, by replacing the integration of molecular systems over time with the training of generative neural networks. This neural network approach to MD samples rare events at a higher rate than traditional MD, however critical gaps in the theory and computational feasibility of Boltzmann generators significantly reduce their usability. Here, we develop a mathematical foundation to overcome these barriers; we demonstrate that the Boltzmann generator approach is sufficiently rapid to replace traditional MD for complex macromolecules, such as proteins in specific applications, and we provide a comprehensive toolkit for the exploration of molecular energy landscapes with neural networks.
Drug Discovery Approaches using Quantum Machine Learning
Apr 01, 2021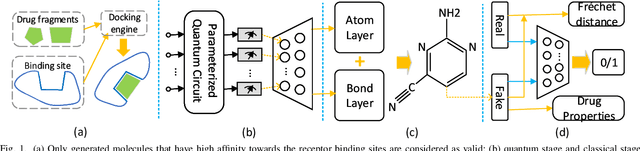
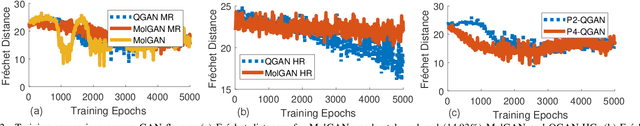
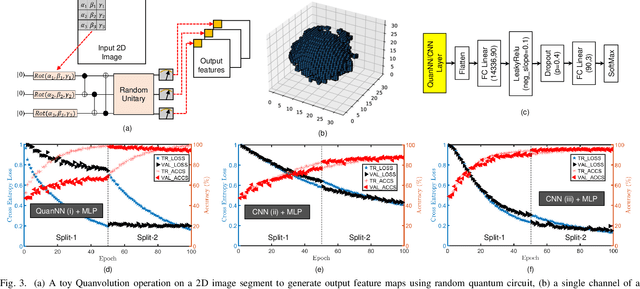
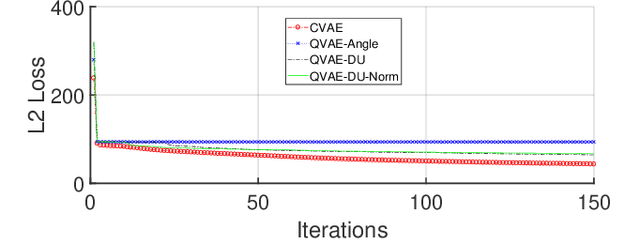
Traditional drug discovery pipeline takes several years and cost billions of dollars. Deep generative and predictive models are widely adopted to assist in drug development. Classical machines cannot efficiently produce atypical patterns of quantum computers which might improve the training quality of learning tasks. We propose a suite of quantum machine learning techniques e.g., generative adversarial network (GAN), convolutional neural network (CNN) and variational auto-encoder (VAE) to generate small drug molecules, classify binding pockets in proteins, and generate large drug molecules, respectively.