 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA comprehensive survey on computational learning methods for analysis of gene expression data in genomics
Feb 16, 2022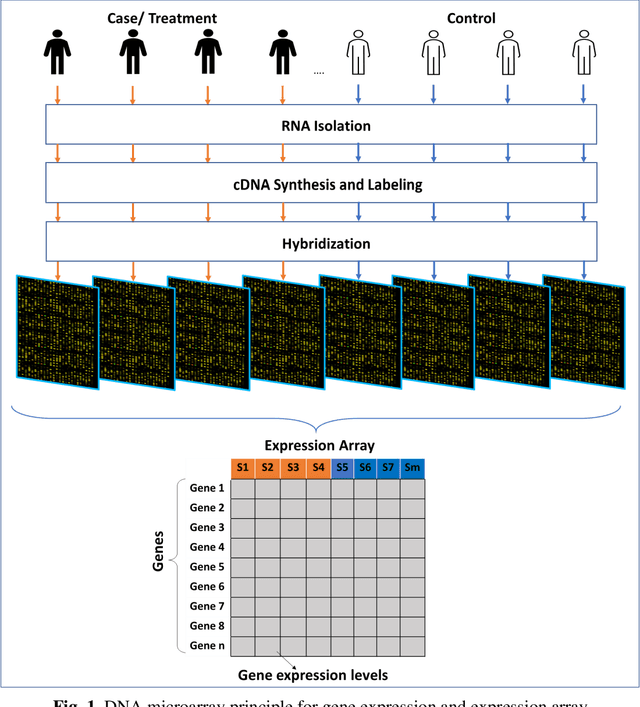

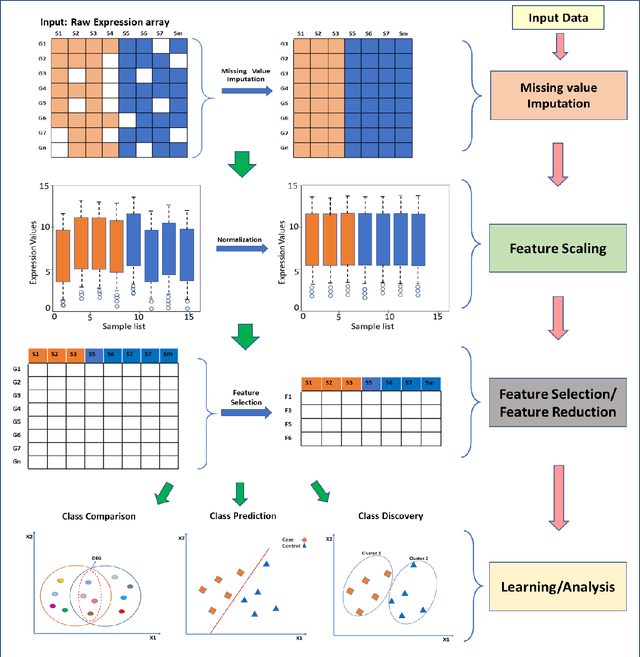
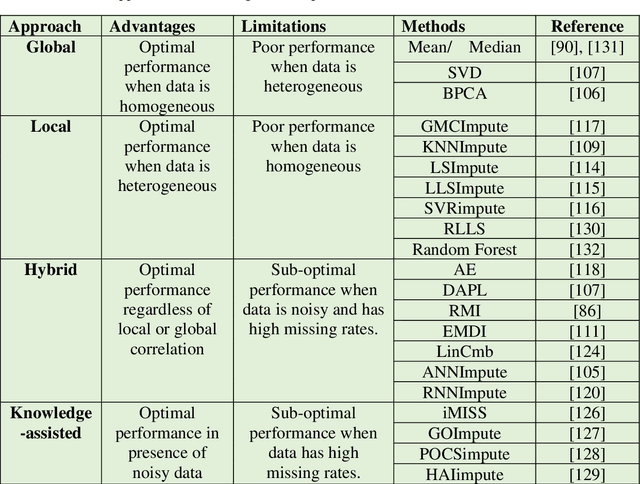
Computational analysis methods including machine learning have a significant impact in the fields of genomics and medicine. High-throughput gene expression analysis methods such as microarray technology and RNA sequencing produce enormous amounts of data. Traditionally, statistical methods are used for comparative analysis of the gene expression data. However, more complex analysis for classification and discovery of feature genes or sample observations requires sophisticated computational approaches. In this review, we compile various statistical and computational tools used in analysis of expression microarray data. Even though, the methods are discussed in the context of expression microarray data, they can also be applied for the analysis of RNA sequencing or quantitative proteomics datasets. We specifically discuss methods for missing value (gene expression) imputation, feature gene scaling, selection and extraction of features for dimensionality reduction, and learning and analysis of expression data. We discuss the types of missing values and the methods and approaches usually employed in their imputation. We also discuss methods of data transformation and feature scaling viz. normalization and standardization. Various approaches used in feature selection and extraction are also reviewed. Lastly, learning and analysis methods including class comparison, class prediction, and class discovery along with their evaluation parameters are described in detail. We have described the process of generation of a microarray gene expression data along with advantages and limitations of the above-mentioned techniques. We believe that this detailed review will help the users to select appropriate methods based on the type of data and the expected outcome.
Comparison of machine learning and deep learning techniques in promoter prediction across diverse species
May 17, 2021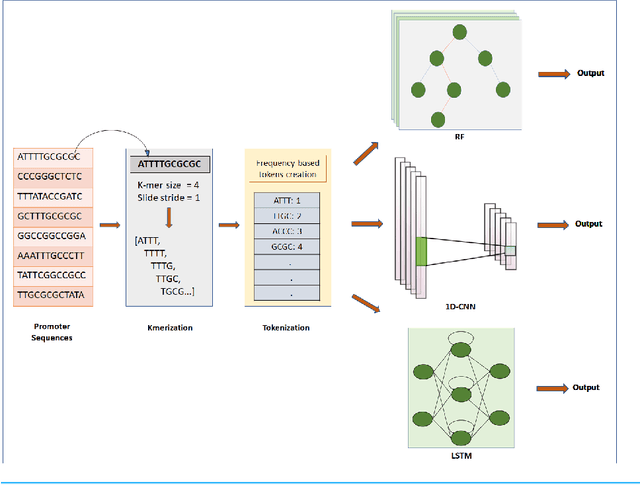

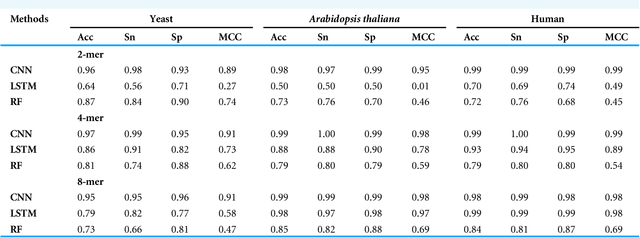

Gene promoters are the key DNA regulatory elements positioned around the transcription start sites and are responsible for regulating gene transcription process. Various alignment-based, signal-based and content-based approaches are reported for the prediction of promoters. However, since all promoter sequences do not show explicit features, the prediction performance of these techniques is poor. Therefore, many machine learning and deep learning models have been proposed for promoter prediction. In this work, we studied methods for vector encoding and promoter classification using genome sequences of three distinct higher eukaryotes viz. yeast (Saccharomyces cerevisiae), A. thaliana (plant) and human (Homo sapiens). We compared one-hot vector encoding method with frequency-based tokenization (FBT) for data pre-processing on 1-D Convolutional Neural Network (CNN) model. We found that FBT gives a shorter input dimension reducing the training time without affecting the sensitivity and specificity of classification. We employed the deep learning techniques, mainly CNN and recurrent neural network with Long Short Term Memory (LSTM) and random forest (RF) classifier for promoter classification at k-mer sizes of 2, 4 and 8. We found CNN to be superior in classification of promoters from non-promoter sequences (binary classification) as well as species-specific classification of promoter sequences (multiclass classification). In summary, the contribution of this work lies in the use of synthetic shuffled negative dataset and frequency-based tokenization for pre-processing. This study provides a comprehensive and generic framework for classification tasks in genomic applications and can be extended to various classification problems.
* 17 pages, 4 figures, 4 tables