 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOne-shot screening of potential peptide ligands on HR1 domain in COVID-19 glycosylated spike (S) protein with deep siamese network
Apr 11, 2020



The novel coronavirus (2019-nCoV) has been declared to be a new international health emergence and no specific drug has been yet identified. Several methods are currently being evaluated such as protease and glycosylated spike (S) protein inhibitors, that outlines the main fusion site among coronavirus and host cells. Notwithstanding, the Heptad Repeat 1 (HR1) domain on the glycosylated spike (S) protein is the region with less mutability and then the most encouraging target for new inhibitors drugs.The novelty of the proposed approach, compared to others, lies in a precise training of a deep neural network toward the 2019-nCoV virus. Where a Siamese Neural Network (SNN) has been trained to distingue the whole 2019-nCoV protein sequence amongst two different viruses family such as HIV-1 and Ebola. In this way, the present deep learning system has precise knowledge of peptide linkage among 2019-nCoV protein structure and differently, of other works, is not trivially trained on public datasets that have not been provided any ligand-peptide information for 2019-nCoV. Suddenly, the SNN shows a sensitivity of $83\%$ of peptide affinity classification, where $3027$ peptides on SATPdb bank have been tested towards the specific region HR1 of 2019-nCoV exhibiting an affinity of $93\%$ for the peptidyl-prolyl cis-trans isomerase (PPIase) peptide. This affinity between PPIase and HR1 can open new horizons of research since several scientific papers have already shown that CsA immunosuppression drug, a main inhibitor of PPIase, suppress the reproduction of different CoV virus included SARS-CoV and MERS-CoV. Finally, to ensure the scientific reproducibility, code and data have been made public at the following link: https://github.com/bionick87/2019-nCoV
Joint analysis of clinical risk factors and 4D cardiac motion for survival prediction using a hybrid deep learning network
Oct 07, 2019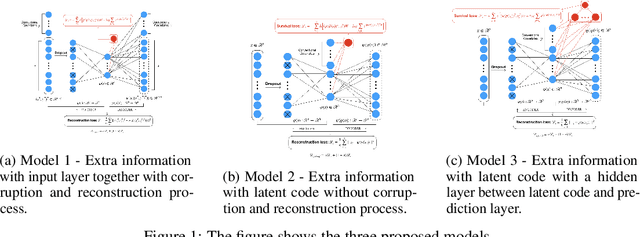
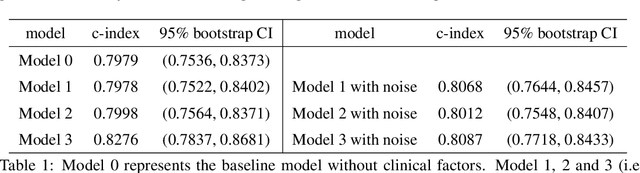
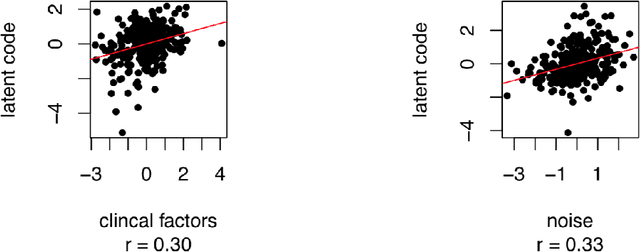
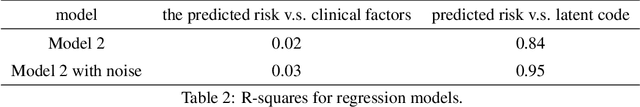
In this work, a novel approach is proposed for joint analysis of high dimensional time-resolved cardiac motion features obtained from segmented cardiac MRI and low dimensional clinical risk factors to improve survival prediction in heart failure. Different methods are evaluated to find the optimal way to insert conventional covariates into deep prediction networks. Correlation analysis between autoencoder latent codes and covariate features is used to examine how these predictors interact. We believe that similar approaches could also be used to introduce knowledge of genetic variants to such survival networks to improve outcome prediction by jointly analysing cardiac motion traits with inheritable risk factors.
* 4 pages, 2 figures