 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-supervised learning for crystal property prediction via denoising
Aug 30, 2024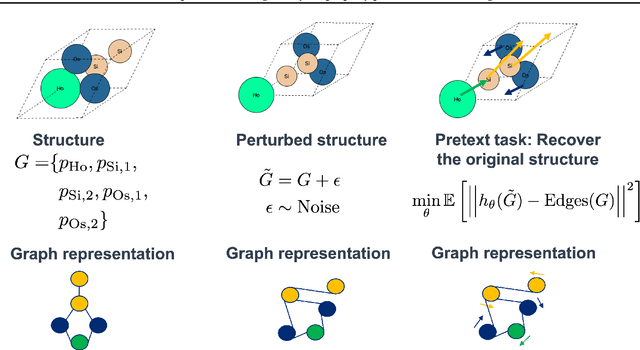
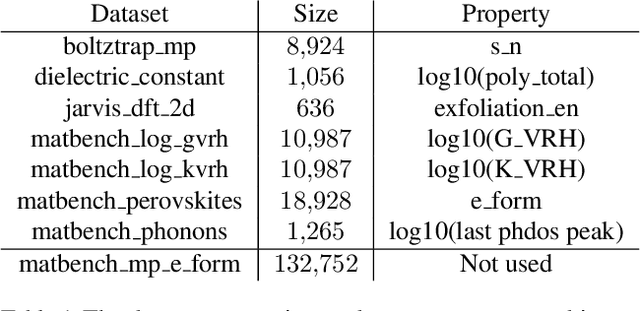
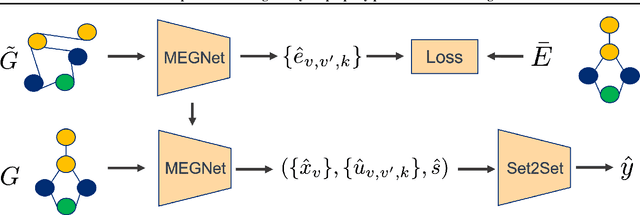

Accurate prediction of the properties of crystalline materials is crucial for targeted discovery, and this prediction is increasingly done with data-driven models. However, for many properties of interest, the number of materials for which a specific property has been determined is much smaller than the number of known materials. To overcome this disparity, we propose a novel self-supervised learning (SSL) strategy for material property prediction. Our approach, crystal denoising self-supervised learning (CDSSL), pretrains predictive models (e.g., graph networks) with a pretext task based on recovering valid material structures when given perturbed versions of these structures. We demonstrate that CDSSL models out-perform models trained without SSL, across material types, properties, and dataset sizes.
Curvature-informed multi-task learning for graph networks
Aug 02, 2022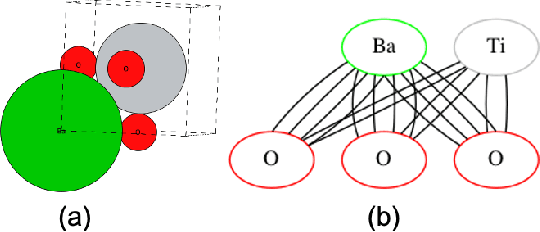
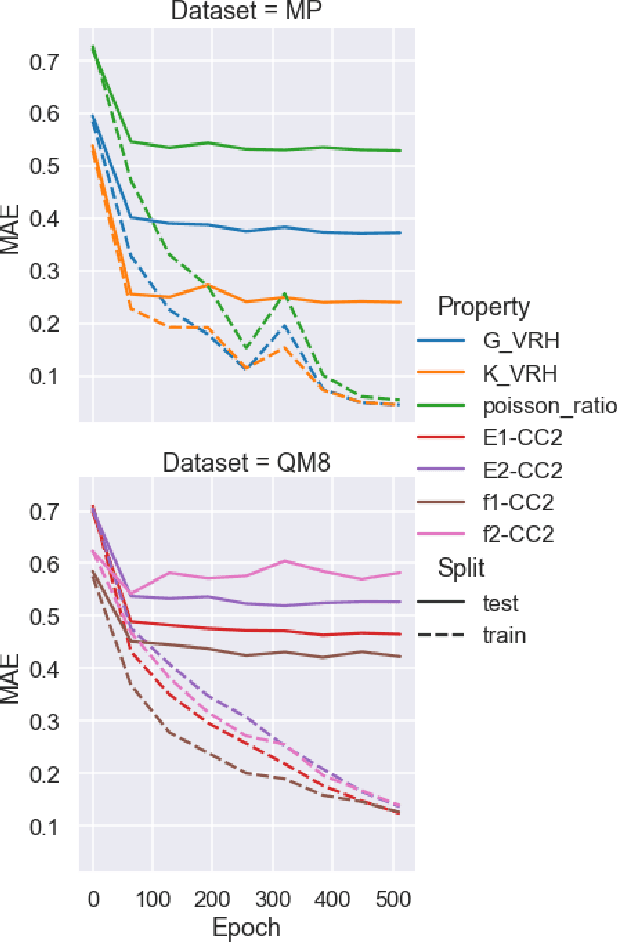
Properties of interest for crystals and molecules, such as band gap, elasticity, and solubility, are generally related to each other: they are governed by the same underlying laws of physics. However, when state-of-the-art graph neural networks attempt to predict multiple properties simultaneously (the multi-task learning (MTL) setting), they frequently underperform a suite of single property predictors. This suggests graph networks may not be fully leveraging these underlying similarities. Here we investigate a potential explanation for this phenomenon: the curvature of each property's loss surface significantly varies, leading to inefficient learning. This difference in curvature can be assessed by looking at spectral properties of the Hessians of each property's loss function, which is done in a matrix-free manner via randomized numerical linear algebra. We evaluate our hypothesis on two benchmark datasets (Materials Project (MP) and QM8) and consider how these findings can inform the training of novel multi-task learning models.