 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLaCoGSEA: Unsupervised deep learning for pathway analysis via latent correlation
Jan 27, 2026Motivation: Pathway enrichment analysis is widely used to interpret gene expression data. Standard approaches, such as GSEA, rely on predefined phenotypic labels and pairwise comparisons, which limits their applicability in unsupervised settings. Existing unsupervised extensions, including single-sample methods, provide pathway-level summaries but primarily capture linear relationships and do not explicitly model gene-pathway associations. More recently, deep learning models have been explored to capture non-linear transcriptomic structure. However, their interpretation has typically relied on generic explainable AI (XAI) techniques designed for feature-level attribution. As these methods are not designed for pathway-level interpretation in unsupervised transcriptomic analyses, their effectiveness in this setting remains limited. Results: To bridge this gap, we introduce LaCoGSEA (Latent Correlation GSEA), an unsupervised framework that integrates deep representation learning with robust pathway statistics. LaCoGSEA employs an autoencoder to capture non-linear manifolds and proposes a global gene-latent correlation metric as a proxy for differential expression, generating dense gene rankings without prior labels. We demonstrate that LaCoGSEA offers three key advantages: (i) it achieves improved clustering performance in distinguishing cancer subtypes compared to existing unsupervised baselines; (ii) it recovers a broader range of biologically meaningful pathways at higher ranks compared with linear dimensionality reduction and gradient-based XAI methods; and (iii) it maintains high robustness and consistency across varying experimental protocols and dataset sizes. Overall, LaCoGSEA provides state-of-the-art performance in unsupervised pathway enrichment analysis. Availability and implementation: https://github.com/willyzzz/LaCoGSEA
TwinPurify: Purifying gene expression data to reveal tumor-intrinsic transcriptional programs via self-supervised learning
Jan 27, 2026Advances in single-cell and spatial transcriptomic technologies have transformed tumor ecosystem profiling at cellular resolution. However, large scale studies on patient cohorts continue to rely on bulk transcriptomic data, where variation in tumor purity obscures tumor-intrinsic transcriptional signals and constrains downstream discovery. Many deconvolution methods report strong performance on synthetic bulk mixtures but fail to generalize to real patient cohorts because of unmodeled biological and technical variation. Here, we introduce TwinPurify, a representation learning framework that adapts the Barlow Twins self-supervised objective, representing a fundamental departure from the deconvolution paradigm. Rather than resolving the bulk mixture into discrete cell-type fractions, TwinPurify instead learns continuous, high-dimensional tumor embeddings by leveraging adjacent-normal profiles within the same cohort as "background" guidance, enabling the disentanglement of tumor-specific signals without relying on any external reference. Benchmarked against multiple large cancer cohorts across RNA-seq and microarray platforms, TwinPurify outperforms conventional representation learning baselines like auto-encoders in recovering tumor-intrinsic and immune signals. The purified embeddings improve molecular subtype and grade classification, enhance survival model concordance, and uncover biologically meaningful pathway activities compared to raw bulk profiles. By providing a transferable framework for decontaminating bulk transcriptomics, TwinPurify extends the utility of existing clinical datasets for molecular discovery.
Can We Edit LLMs for Long-Tail Biomedical Knowledge?
Apr 14, 2025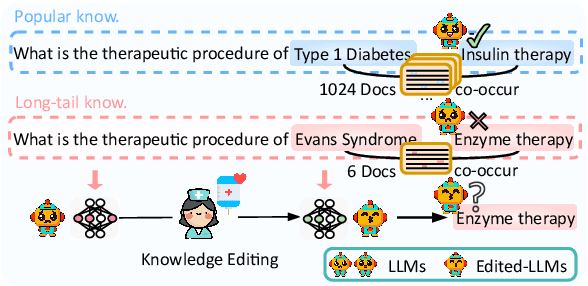
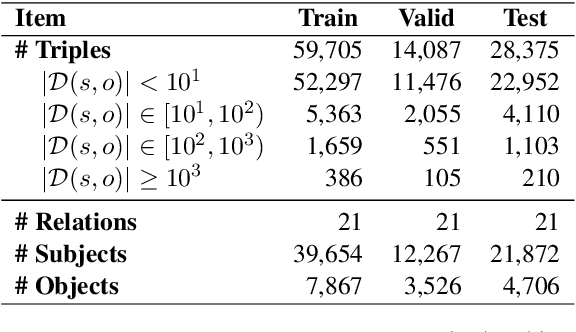
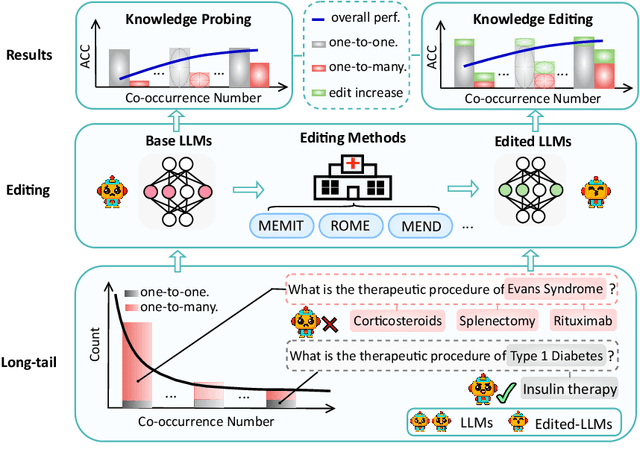
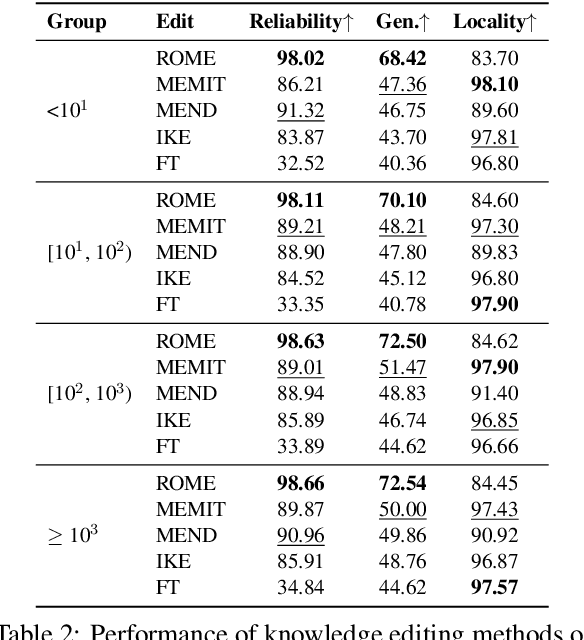
Knowledge editing has emerged as an effective approach for updating large language models (LLMs) by modifying their internal knowledge. However, their application to the biomedical domain faces unique challenges due to the long-tailed distribution of biomedical knowledge, where rare and infrequent information is prevalent. In this paper, we conduct the first comprehensive study to investigate the effectiveness of knowledge editing methods for editing long-tail biomedical knowledge. Our results indicate that, while existing editing methods can enhance LLMs' performance on long-tail biomedical knowledge, their performance on long-tail knowledge remains inferior to that on high-frequency popular knowledge, even after editing. Our further analysis reveals that long-tail biomedical knowledge contains a significant amount of one-to-many knowledge, where one subject and relation link to multiple objects. This high prevalence of one-to-many knowledge limits the effectiveness of knowledge editing in improving LLMs' understanding of long-tail biomedical knowledge, highlighting the need for tailored strategies to bridge this performance gap.
Generalized Categorisation of Digital Pathology Whole Image Slides using Unsupervised Learning
Dec 27, 2020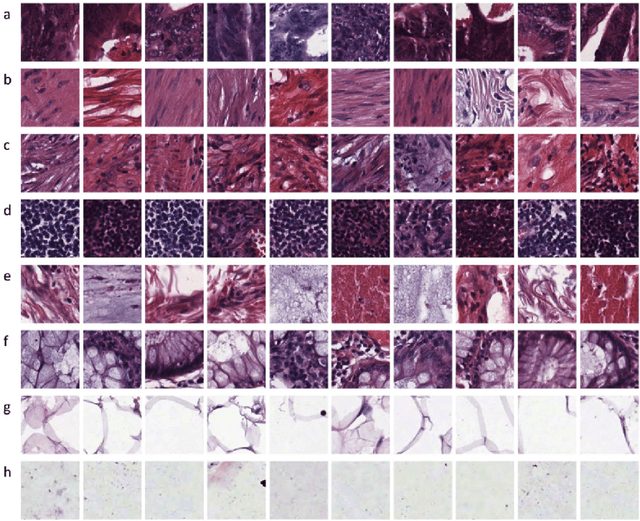
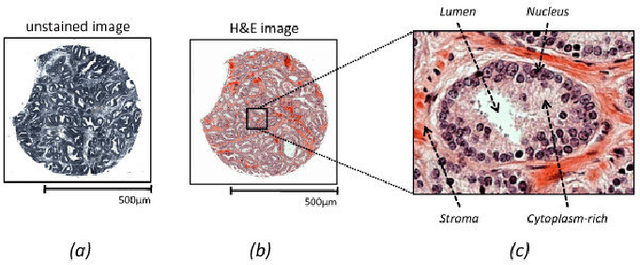
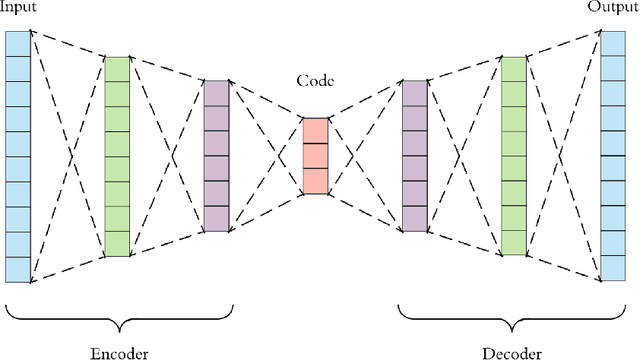
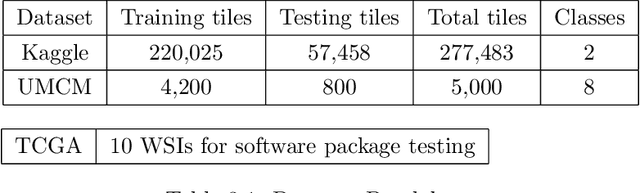
This project aims to break down large pathology images into small tiles and then cluster those tiles into distinct groups without the knowledge of true labels, our analysis shows how difficult certain aspects of clustering tumorous and non-tumorous cells can be and also shows that comparing the results of different unsupervised approaches is not a trivial task. The project also provides a software package to be used by the digital pathology community, that uses some of the approaches developed to perform unsupervised unsupervised tile classification, which could then be easily manually labelled. The project uses a mixture of techniques ranging from classical clustering algorithms such as K-Means and Gaussian Mixture Models to more complicated feature extraction techniques such as deep Autoencoders and Multi-loss learning. Throughout the project, we attempt to set a benchmark for evaluation using a few measures such as completeness scores and cluster plots. Throughout our results we show that Convolutional Autoencoders manages to slightly outperform the rest of the approaches due to its powerful internal representation learning abilities. Moreover, we show that Gaussian Mixture models produce better results than K-Means on average due to its flexibility in capturing different clusters. We also show the huge difference in the difficulties of classifying different types of pathology textures.