 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Networks embedded into Margules model for vapor-liquid equilibria prediction
Feb 26, 2025



Predictive thermodynamic models are crucial for the early stages of product and process design. In this paper the performance of Graph Neural Networks (GNNs) embedded into a relatively simple excess Gibbs energy model, the extended Margules model, for predicting vapor-liquid equilibrium is analyzed. By comparing its performance against the established UNIFAC-Dortmund model it has been shown that GNNs embedded in Margules achieves an overall lower accuracy. However, higher accuracy is observed in the case of various types of binary mixtures. Moreover, since group contribution methods, like UNIFAC, are limited due to feasibility of molecular fragmentation or availability of parameters, the GNN in Margules model offers an alternative for VLE estimation. The findings establish a baseline for the predictive accuracy that simple excess Gibbs energy models combined with GNNs trained solely on infinite dilution data can achieve.
An analysis of optimization problems involving ReLU neural networks
Feb 05, 2025



Solving mixed-integer optimization problems with embedded neural networks with ReLU activation functions is challenging. Big-M coefficients that arise in relaxing binary decisions related to these functions grow exponentially with the number of layers. We survey and propose different approaches to analyze and improve the run time behavior of mixed-integer programming solvers in this context. Among them are clipped variants and regularization techniques applied during training as well as optimization-based bound tightening and a novel scaling for given ReLU networks. We numerically compare these approaches for three benchmark problems from the literature. We use the number of linear regions, the percentage of stable neurons, and overall computational effort as indicators. As a major takeaway we observe and quantify a trade-off between the often desired redundancy of neural network models versus the computational costs for solving related optimization problems.
Learning reduced-order Quadratic-Linear models in Process Engineering using Operator Inference
Feb 27, 2024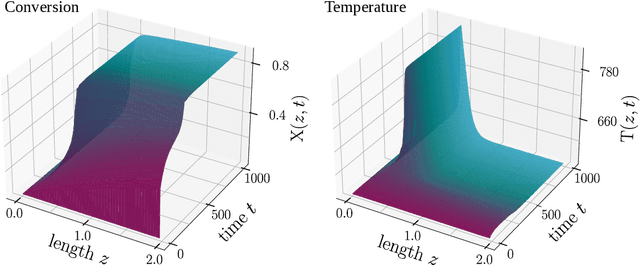
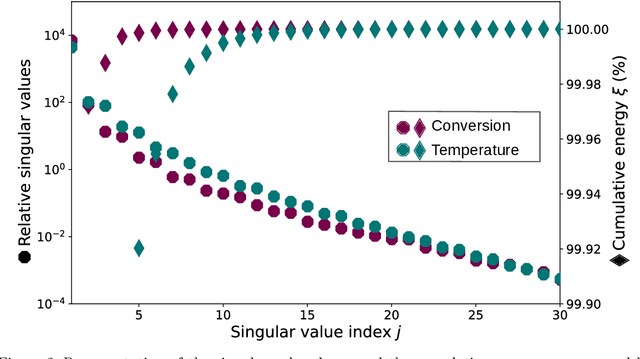

In this work, we address the challenge of efficiently modeling dynamical systems in process engineering. We use reduced-order model learning, specifically operator inference. This is a non-intrusive, data-driven method for learning dynamical systems from time-domain data. The application in our study is carbon dioxide methanation, an important reaction within the Power-to-X framework, to demonstrate its potential. The numerical results show the ability of the reduced-order models constructed with operator inference to provide a reduced yet accurate surrogate solution. This represents an important milestone towards the implementation of fast and reliable digital twin architectures.
Gibbs-Helmholtz Graph Neural Network: capturing the temperature dependency of activity coefficients at infinite dilution
Dec 16, 2022



The accurate prediction of physicochemical properties of chemical compounds in mixtures (such as the activity coefficient at infinite dilution $\gamma_{ij}^\infty$) is essential for developing novel and more sustainable chemical processes. In this work, we analyze the performance of previously-proposed GNN-based models for the prediction of $\gamma_{ij}^\infty$, and compare them with several mechanistic models in a series of 9 isothermal studies. Moreover, we develop the Gibbs-Helmholtz Graph Neural Network (GH-GNN) model for predicting $\ln \gamma_{ij}^\infty$ of molecular systems at different temperatures. Our method combines the simplicity of a Gibbs-Helmholtz-derived expression with a series of graph neural networks that incorporate explicit molecular and intermolecular descriptors for capturing dispersion and hydrogen bonding effects. We have trained this model using experimentally determined $\ln \gamma_{ij}^\infty$ data of 40,219 binary-systems involving 1032 solutes and 866 solvents, overall showing superior performance compared to the popular UNIFAC-Dortmund model. We analyze the performance of GH-GNN for continuous and discrete inter/extrapolation and give indications for the model's applicability domain and expected accuracy. In general, GH-GNN is able to produce accurate predictions for extrapolated binary-systems if at least 25 systems with the same combination of solute-solvent chemical classes are contained in the training set and a similarity indicator above 0.35 is also present. This model and its applicability domain recommendations have been made open-source at https://github.com/edgarsmdn/GH-GNN.