 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLncRNA-disease association prediction method based on heterogeneous information completion and convolutional neural network
Jun 02, 2024



The emerging research shows that lncRNA has crucial research value in a series of complex human diseases. Therefore, the accurate identification of lncRNA-disease associations (LDAs) is very important for the warning and treatment of diseases. However, most of the existing methods have limitations in identifying nonlinear LDAs, and it remains a huge challenge to predict new LDAs. In this paper, a deep learning model based on a heterogeneous network and convolutional neural network (CNN) is proposed for lncRNA-disease association prediction, named HCNNLDA. The heterogeneous network containing the lncRNA, disease, and miRNA nodes, is constructed firstly. The embedding matrix of a lncRNA-disease node pair is constructed according to various biological premises about lncRNAs, diseases, and miRNAs. Then, the low-dimensional feature representation is fully learned by the convolutional neural network. In the end, the XGBoot classifier model is trained to predict the potential LDAs. HCNNLDA obtains a high AUC value of 0.9752 and AUPR of 0.9740 under the 5-fold cross-validation. The experimental results show that the proposed model has better performance than that of several latest prediction models. Meanwhile, the effectiveness of HCNNLDA in identifying novel LDAs is further demonstrated by case studies of three diseases. To sum up, HCNNLDA is a feasible calculation model to predict LDAs.
Heterogeneous network and graph attention auto-encoder for LncRNA-disease association prediction
May 03, 2024

The emerging research shows that lncRNAs are associated with a series of complex human diseases. However, most of the existing methods have limitations in identifying nonlinear lncRNA-disease associations (LDAs), and it remains a huge challenge to predict new LDAs. Therefore, the accurate identification of LDAs is very important for the warning and treatment of diseases. In this work, multiple sources of biomedical data are fully utilized to construct characteristics of lncRNAs and diseases, and linear and nonlinear characteristics are effectively integrated. Furthermore, a novel deep learning model based on graph attention automatic encoder is proposed, called HGATELDA. To begin with, the linear characteristics of lncRNAs and diseases are created by the miRNA-lncRNA interaction matrix and miRNA-disease interaction matrix. Following this, the nonlinear features of diseases and lncRNAs are extracted using a graph attention auto-encoder, which largely retains the critical information and effectively aggregates the neighborhood information of nodes. In the end, LDAs can be predicted by fusing the linear and nonlinear characteristics of diseases and lncRNA. The HGATELDA model achieves an impressive AUC value of 0.9692 when evaluated using a 5-fold cross-validation indicating its superior performance in comparison to several recent prediction models. Meanwhile, the effectiveness of HGATELDA in identifying novel LDAs is further demonstrated by case studies. the HGATELDA model appears to be a viable computational model for predicting LDAs.
Supervised Discriminative Sparse PCA for Com-Characteristic Gene Selection and Tumor Classification on Multiview Biological Data
May 28, 2019
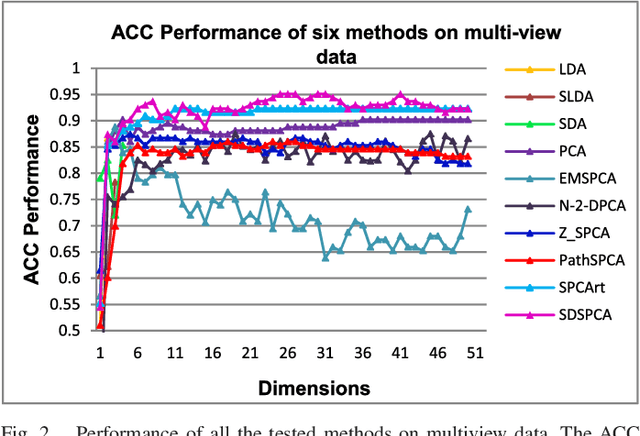


Principal Component Analysis (PCA) has been used to study the pathogenesis of diseases. To enhance the interpretability of classical PCA, various improved PCA methods have been proposed to date. Among these, a typical method is the so-called sparse PCA, which focuses on seeking sparse loadings. However, the performance of these methods is still far from satisfactory due to their limitation of using unsupervised learning methods; moreover, the class ambiguity within the sample is high. To overcome this problem, this study developed a new PCA method, which is named the Supervised Discriminative Sparse PCA (SDSPCA). The main innovation of this method is the incorporation of discriminative information and sparsity into the PCA model. Specifically, in contrast to the traditional sparse PCA, which imposes sparsity on the loadings, here, sparse components are obtained to represent the data. Furthermore, via linear transformation, the sparse components approximate the given label information. On the one hand, sparse components improve interpretability over traditional PCA, while on the other hand, they are have discriminative abilities suitable for classification purposes. A simple algorithm is developed and its convergence proof is provided. The SDSPCA has been applied to common characteristic gene selection (com-characteristic gene) and tumor classification on multi-view biological data. The sparsity and classification performance of the SDSPCA are empirically verified via abundant, reasonable, and effective experiments, and the obtained results demonstrate that SDSPCA outperforms other state-of-the-art methods.