 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground
Aug 23, 2019

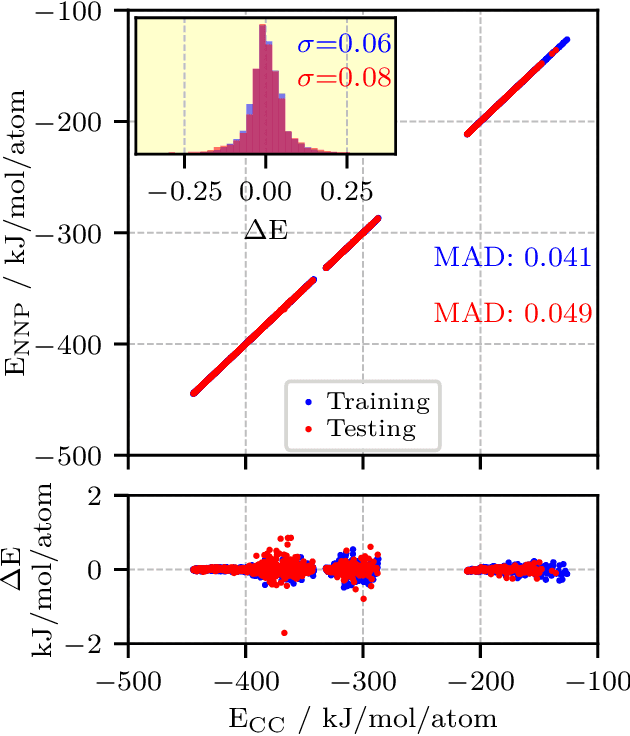

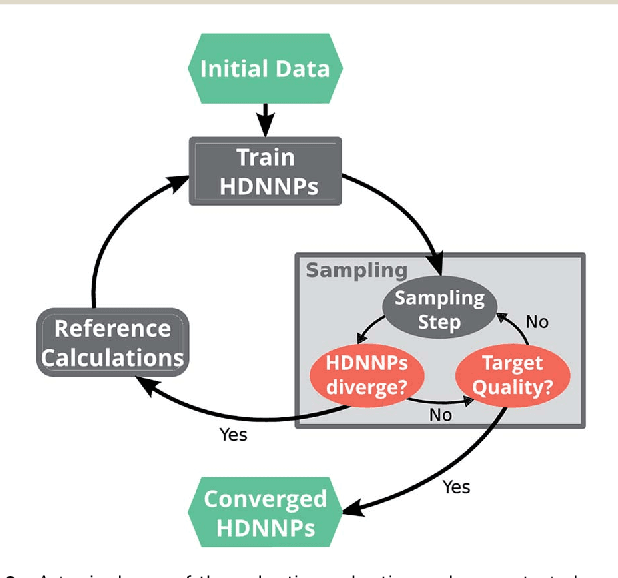
Highly accurate potential energy surfaces are of key interest for the detailed understanding and predictive modeling of chemical systems. In recent years, several new types of force fields, which are based on machine learning algorithms and fitted to ab initio reference calculations, have been introduced to meet this requirement. Here we show how high-dimensional neural network potentials can be employed to automatically generate the potential energy surface of finite sized clusters at coupled cluster accuracy, namely CCSD(T*)-F12a/aug-cc-pVTZ. The developed automated procedure utilizes the established intrinsic properties of the model such that the configurations for the training set are selected in an unbiased and efficient way to minimize the computational effort of expensive reference calculations. These ideas are applied to protonated water clusters from the hydronium cation, H$_3$O$^+$, up to the tetramer, H$_9$O$_{4}^{+}$, and lead to a common potential energy surface that describes all these systems at essentially converged coupled cluster accuracy with a fitting error of 0.06 kJ/mol per atom. The fit is validated in detail and separately for all clusters up to the tetramer and yields reliable results not only for stationary points, but also for reaction pathways, intermediate configurations, as well as different sampling techniques. Per design the NNPs constructed in this fashion can handle very different conditions including the quantum nature of the nuclei and enhanced sampling techniques covering very low as well as high temperatures. This enables fast and exhaustive exploration of the targeted protonated water clusters at essentially converged interactions. In addition, the automated process will allow one to tackle finite systems much beyond the present case.
Machine Learning Molecular Dynamics for the Simulation of Infrared Spectra
May 16, 2017


Machine learning has emerged as an invaluable tool in many research areas. In the present work, we harness this power to predict highly accurate molecular infrared spectra with unprecedented computational efficiency. To account for vibrational anharmonic and dynamical effects -- typically neglected by conventional quantum chemistry approaches -- we base our machine learning strategy on ab initio molecular dynamics simulations. While these simulations are usually extremely time consuming even for small molecules, we overcome these limitations by leveraging the power of a variety of machine learning techniques, not only accelerating simulations by several orders of magnitude, but also greatly extending the size of systems that can be treated. To this end, we develop a molecular dipole moment model based on environment dependent neural network charges and combine it with the neural network potentials of Behler and Parrinello. Contrary to the prevalent big data philosophy, we are able to obtain very accurate machine learning models for the prediction of infrared spectra based on only a few hundreds of electronic structure reference points. This is made possible through the introduction of a fully automated sampling scheme and the use of molecular forces during neural network potential training. We demonstrate the power of our machine learning approach by applying it to model the infrared spectra of a methanol molecule, n-alkanes containing up to 200 atoms and the protonated alanine tripeptide, which at the same time represents the first application of machine learning techniques to simulate the dynamics of a peptide. In all these case studies we find excellent agreement between the infrared spectra predicted via machine learning models and the respective theoretical and experimental spectra.