 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlexible Kernels for Protein Property Prediction
Jun 09, 2026Despite its importance to applications in protein design, predicting protein properties like binding affinity and thermostability from sparse experimental data remains a significant challenge. Accordingly, we introduce a class of sequence kernels that exploit evolutionary substitution matrices as well as local linearity and demonstrate that the resulting Gaussian processes provide data-efficient models of protein property landscapes, frequently outperforming alternatives that rely on foundation model embeddings. Furthermore--by learning what are in effect structure-aware substitution matrices--we show that our kernels can readily incorporate structural information from foundation models. We demonstrate that these structure-conditioned kernels are well suited to multi-task learning across multiple protein property landscapes and can decisively outperform local supervised learning methods.
Guide your favorite protein sequence generative model
May 07, 2025Generative machine learning models have begun to transform protein engineering, yet no principled framework for conditioning on auxiliary information in a plug-and-play manner exists; one may want to iteratively incorporate experimental feedback, or make use of an existing classifier -- such as for predicting enzyme commission number -- in order to guide the sampling of the generative model to generate sequences with desired properties. Herein, we present ProteinGuide, a rigorous and general framework to achieve just that: through unifying a broad class of protein generative models that includes masked language, (order-agnostic) autoregressive, diffusion and flow-matching models, we provide an approach to statistically condition pre-trained protein generative models. We demonstrate applicability of our approach by guiding each of two commonly used protein generative models, ProteinMPNN and ESM3, to generate amino acid and structure token sequences conditioned on several user-specified properties, namely, enhanced stability and CATH-labeled fold generation.
Unlocking Guidance for Discrete State-Space Diffusion and Flow Models
Jun 03, 2024Generative models on discrete state-spaces have a wide range of potential applications, particularly in the domain of natural sciences. In continuous state-spaces, controllable and flexible generation of samples with desired properties has been realized using guidance on diffusion and flow models. However, these guidance approaches are not readily amenable to discrete state-space models. Consequently, we introduce a general and principled method for applying guidance on such models. Our method depends on leveraging continuous-time Markov processes on discrete state-spaces, which unlocks computational tractability for sampling from a desired guided distribution. We demonstrate the utility of our approach, Discrete Guidance, on a range of applications including guided generation of images, small-molecules, DNA sequences and protein sequences.
Augmenting Neural Networks with Priors on Function Values
Feb 21, 2022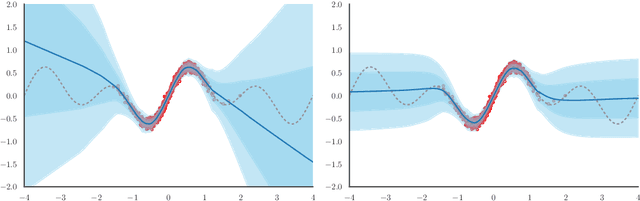



The need for function estimation in label-limited settings is common in the natural sciences. At the same time, prior knowledge of function values is often available in these domains. For example, data-free biophysics-based models can be informative on protein properties, while quantum-based computations can be informative on small molecule properties. How can we coherently leverage such prior knowledge to help improve a neural network model that is quite accurate in some regions of input space -- typically near the training data -- but wildly wrong in other regions? Bayesian neural networks (BNN) enable the user to specify prior information only on the neural network weights, not directly on the function values. Moreover, there is in general no clear mapping between these. Herein, we tackle this problem by developing an approach to augment BNNs with prior information on the function values themselves. Our probabilistic approach yields predictions that rely more heavily on the prior information when the epistemic uncertainty is large, and more heavily on the neural network when the epistemic uncertainty is small.
A deep-learning view of chemical space designed to facilitate drug discovery
Feb 07, 2020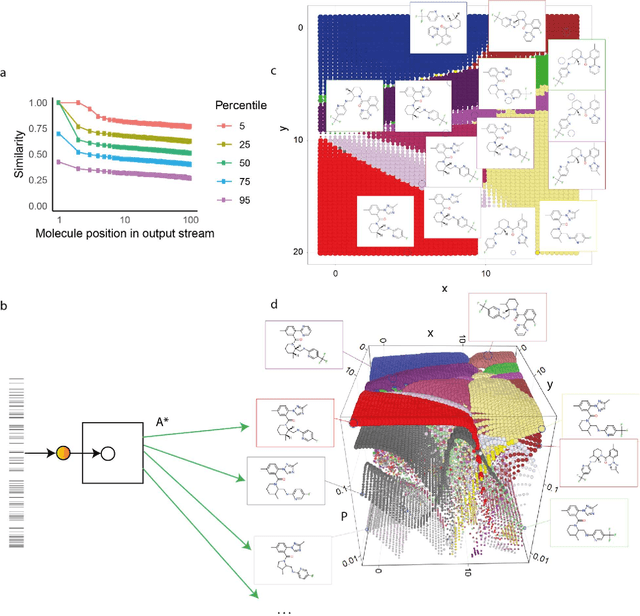
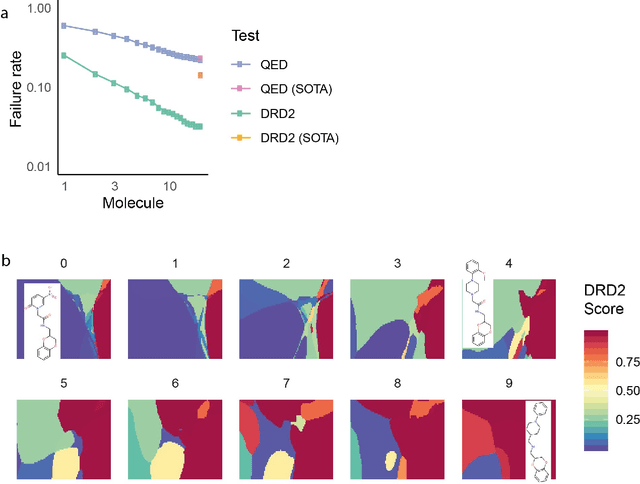
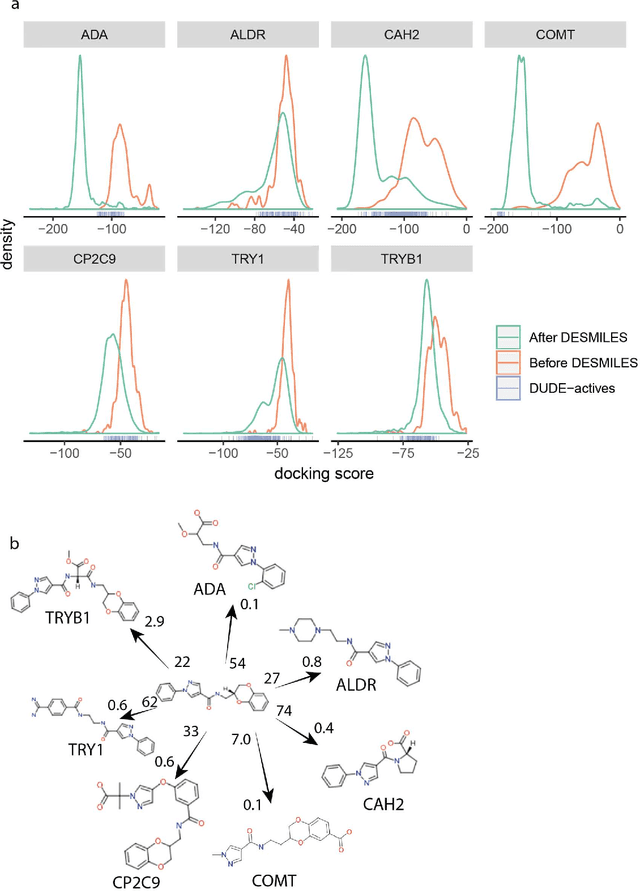
Drug discovery projects entail cycles of design, synthesis, and testing that yield a series of chemically related small molecules whose properties, such as binding affinity to a given target protein, are progressively tailored to a particular drug discovery goal. The use of deep learning technologies could augment the typical practice of using human intuition in the design cycle, and thereby expedite drug discovery projects. Here we present DESMILES, a deep neural network model that advances the state of the art in machine learning approaches to molecular design. We applied DESMILES to a previously published benchmark that assesses the ability of a method to modify input molecules to inhibit the dopamine receptor D2, and DESMILES yielded a 77% lower failure rate compared to state-of-the-art models. To explain the ability of DESMILES to hone molecular properties, we visualize a layer of the DESMILES network, and further demonstrate this ability by using DESMILES to tailor the same molecules used in the D2 benchmark test to dock more potently against seven different receptors.