 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOn the Trap Space Semantics of Normal Logic Programs
Jan 07, 2026The logical semantics of normal logic programs has traditionally been based on the notions of Clark's completion and two-valued or three-valued canonical models, including supported, stable, regular, and well-founded models. Two-valued interpretations can also be seen as states evolving under a program's update operator, producing a transition graph whose fixed points and cycles capture stable and oscillatory behaviors, respectively. We refer to this view as dynamical semantics since it characterizes the program's meaning in terms of state-space trajectories, as first introduced in the stable (supported) class semantics. Recently, we have established a formal connection between Datalog^\neg programs (i.e., normal logic programs without function symbols) and Boolean networks, leading to the introduction of the trap space concept for Datalog^\neg programs. In this paper, we generalize the trap space concept to arbitrary normal logic programs, introducing trap space semantics as a new approach to their interpretation. This new semantics admits both model-theoretic and dynamical characterizations, providing a comprehensive approach to understanding program behavior. We establish the foundational properties of the trap space semantics and systematically relate it to the established model-theoretic semantics, including the stable (supported), stable (supported) partial, regular, and L-stable model semantics, as well as to the dynamical stable (supported) class semantics. Our results demonstrate that the trap space semantics offers a unified and precise framework for proving the existence of supported classes, strict stable (supported) classes, and regular models, in addition to uncovering and formalizing deeper relationships among the existing semantics of normal logic programs.
* In Proceedings ICLP 2025, arXiv:2601.00047
On the Boolean Network Theory of Datalog$^ eg$
Apr 21, 2025Datalog$^\neg$ is a central formalism used in a variety of domains ranging from deductive databases and abstract argumentation frameworks to answer set programming. Its model theory is the finite counterpart of the logical semantics developed for normal logic programs, mainly based on the notions of Clark's completion and two-valued or three-valued canonical models including supported, stable, regular and well-founded models. In this paper we establish a formal link between Datalog$^\neg$ and Boolean network theory, which was initially introduced by Stuart Kaufman and Ren\'e Thomas to reason about gene regulatory networks. We use previous results from Boolean network theory to prove that in the absence of odd cycles in a Datalog$^\neg$ program, the regular models coincide with the stable models, which entails the existence of stable models, and in the absence of even cycles, we show the uniqueness of stable partial models, which entails the uniqueness of regular models. These results on regular models have been claimed by You and Yuan in 1994 for normal logic programs but we show problems in their definition of well-founded stratification and in their proofs that we can fix for negative normal logic programs only. We also give upper bounds on the numbers of stable partial, regular, and stable models of a Datalog$^\neg$ program using the cardinality of a feedback vertex set in its atom dependency graph. Interestingly, our connection to Boolean network theory also points us to the notion of trap spaces for Datalog$^\neg$ programs. We relate the notions of supported or stable trap spaces to the other semantics of Datalog$^\neg$, and show the equivalence between subset-minimal stable trap spaces and regular models.
Graphical Conditions for the Existence, Unicity and Number of Regular Models
Feb 13, 2025The regular models of a normal logic program are a particular type of partial (i.e. 3-valued) models which correspond to stable partial models with minimal undefinedness. In this paper, we explore graphical conditions on the dependency graph of a finite ground normal logic program to analyze the existence, unicity and number of regular models for the program. We show three main results: 1) a necessary condition for the existence of non-trivial (i.e. non-2-valued) regular models, 2) a sufficient condition for the unicity of regular models, and 3) two upper bounds for the number of regular models based on positive feedback vertex sets. The first two conditions generalize the finite cases of the two existing results obtained by You and Yuan (1994) for normal logic programs with well-founded stratification. The third result is also new to the best of our knowledge. Key to our proofs is a connection that we establish between finite ground normal logic programs and Boolean network theory.
* In Proceedings ICLP 2024, arXiv:2502.08453
A Skin Microbiome Model with AMP interactions and Analysis of Quasi-Stability vs Stability in Population Dynamics
Oct 23, 2023
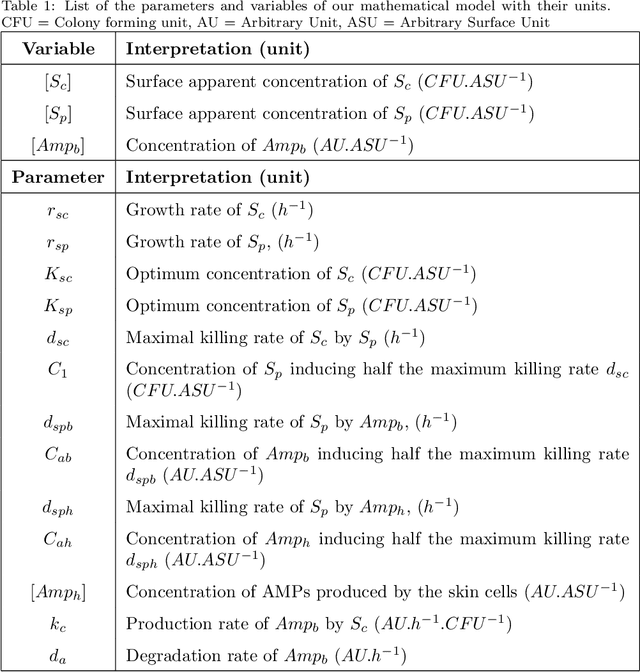


The skin microbiome plays an important role in the maintenance of a healthy skin. It is an ecosystem, composed of several species, competing for resources and interacting with the skin cells. Imbalance in the cutaneous microbiome, also called dysbiosis, has been correlated with several skin conditions, including acne and atopic dermatitis. Generally, dysbiosis is linked to colonization of the skin by a population of opportunistic pathogenic bacteria. Treatments consisting in non-specific elimination of cutaneous microflora have shown conflicting results. In this article, we introduce a mathematical model based on ordinary differential equations, with 2 types of bacteria populations (skin commensals and opportunistic pathogens) and including the production of antimicrobial peptides to study the mechanisms driving the dominance of one population over the other. By using published experimental data, assumed to correspond to the observation of stable states in our model, we reduce the number of parameters of the model from 13 to 5. We then use a formal specification in quantitative temporal logic to calibrate our model by global parameter optimization and perform sensitivity analyses. On the time scale of 2 days of the experiments, the model predicts that certain changes of the environment, like the elevation of skin surface pH, create favorable conditions for the emergence and colonization of the skin by the opportunistic pathogen population, while the production of human AMPs has non-linear effect on the balance between pathogens and commensals. Surprisingly, simulations on longer time scales reveal that the equilibrium reached around 2 days can in fact be a quasi-stable state followed by the reaching of a reversed stable state after 12 days or more. We analyse the conditions of quasi-stability observed in this model using tropical algebraic methods, and show their non-generic character in contrast to slow-fast systems. These conditions are then generalized to a large class of population dynamics models over any number of species.
Neural-based classification rule learning for sequential data
Feb 22, 2023Discovering interpretable patterns for classification of sequential data is of key importance for a variety of fields, ranging from genomics to fraud detection or more generally interpretable decision-making. In this paper, we propose a novel differentiable fully interpretable method to discover both local and global patterns (i.e. catching a relative or absolute temporal dependency) for rule-based binary classification. It consists of a convolutional binary neural network with an interpretable neural filter and a training strategy based on dynamically-enforced sparsity. We demonstrate the validity and usefulness of the approach on synthetic datasets and on an open-source peptides dataset. Key to this end-to-end differentiable method is that the expressive patterns used in the rules are learned alongside the rules themselves.
Reactmine: a search algorithm for inferring chemical reaction networks from time series data
Sep 07, 2022
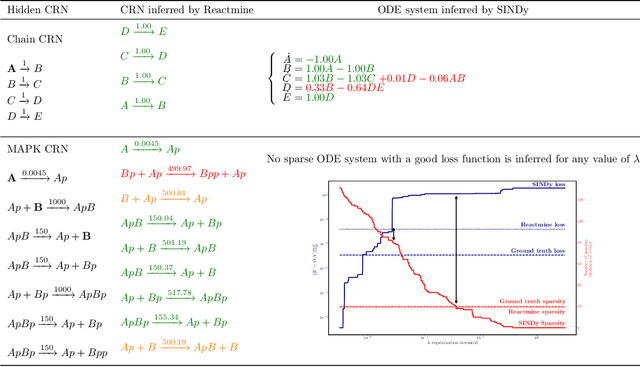
Inferring chemical reaction networks (CRN) from time series data is a challenge encouraged by the growing availability of quantitative temporal data at the cellular level. This motivates the design of algorithms to infer the preponderant reactions between the molecular species observed in a given biochemical process, and help to build CRN model structure and kinetics. Existing ODE-based inference methods such as SINDy resort to least square regression combined with sparsity-enforcing penalization, such as Lasso. However, when the input time series are only available in wild type conditions in which all reactions are present, we observe that current methods fail to learn sparse models. Results: We present Reactmine, a CRN learning algorithm which enforces sparsity by inferring reactions in a sequential fashion within a search tree of bounded depth, ranking the inferred reaction candidates according to the variance of their kinetics, and re-optimizing the CRN kinetic parameters on the whole trace in a final pass to rank the inferred CRN candidates. We first evaluate its performance on simulation data from a benchmark of hidden CRNs, together with algorithmic hyperparameter sensitivity analyses, and then on two sets of real experimental data: one from protein fluorescence videomicroscopy of cell cycle and circadian clock markers, and one from biomedical measurements of systemic circadian biomarkers possibly acting on clock gene expression in peripheral organs. We show that Reactmine succeeds both on simulation data by retrieving hidden CRNs where SINDy fails, and on the two real datasets by inferring reactions in agreement with previous studies.