 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Multi-Objective Bayesian Optimization with Scalable Batch Evaluations for Sample-Efficient De Novo Molecular Design
Dec 19, 2025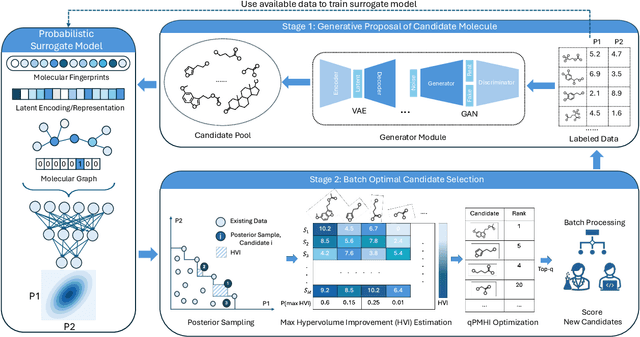
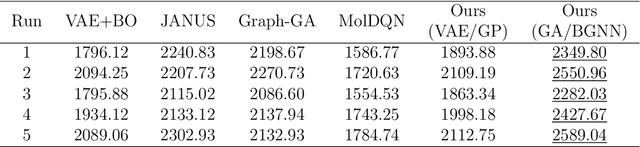
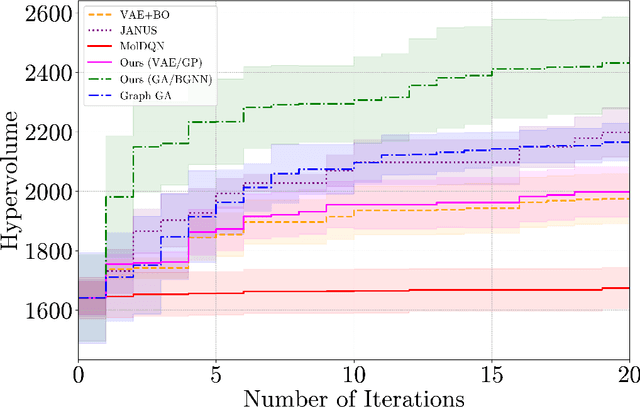

Designing molecules that must satisfy multiple, often conflicting objectives is a central challenge in molecular discovery. The enormous size of chemical space and the cost of high-fidelity simulations have driven the development of machine learning-guided strategies for accelerating design with limited data. Among these, Bayesian optimization (BO) offers a principled framework for sample-efficient search, while generative models provide a mechanism to propose novel, diverse candidates beyond fixed libraries. However, existing methods that couple the two often rely on continuous latent spaces, which introduces both architectural entanglement and scalability challenges. This work introduces an alternative, modular "generate-then-optimize" framework for de novo multi-objective molecular design/discovery. At each iteration, a generative model is used to construct a large, diverse pool of candidate molecules, after which a novel acquisition function, qPMHI (multi-point Probability of Maximum Hypervolume Improvement), is used to optimally select a batch of candidates most likely to induce the largest Pareto front expansion. The key insight is that qPMHI decomposes additively, enabling exact, scalable batch selection via only simple ranking of probabilities that can be easily estimated with Monte Carlo sampling. We benchmark the framework against state-of-the-art latent-space and discrete molecular optimization methods, demonstrating significant improvements across synthetic benchmarks and application-driven tasks. Specifically, in a case study related to sustainable energy storage, we show that our approach quickly uncovers novel, diverse, and high-performing organic (quinone-based) cathode materials for aqueous redox flow battery applications.
SyMANTIC: An Efficient Symbolic Regression Method for Interpretable and Parsimonious Model Discovery in Science and Beyond
Feb 05, 2025



Symbolic regression (SR) is an emerging branch of machine learning focused on discovering simple and interpretable mathematical expressions from data. Although a wide-variety of SR methods have been developed, they often face challenges such as high computational cost, poor scalability with respect to the number of input dimensions, fragility to noise, and an inability to balance accuracy and complexity. This work introduces SyMANTIC, a novel SR algorithm that addresses these challenges. SyMANTIC efficiently identifies (potentially several) low-dimensional descriptors from a large set of candidates (from $\sim 10^5$ to $\sim 10^{10}$ or more) through a unique combination of mutual information-based feature selection, adaptive feature expansion, and recursively applied $\ell_0$-based sparse regression. In addition, it employs an information-theoretic measure to produce an approximate set of Pareto-optimal equations, each offering the best-found accuracy for a given complexity. Furthermore, our open-source implementation of SyMANTIC, built on the PyTorch ecosystem, facilitates easy installation and GPU acceleration. We demonstrate the effectiveness of SyMANTIC across a range of problems, including synthetic examples, scientific benchmarks, real-world material property predictions, and chaotic dynamical system identification from small datasets. Extensive comparisons show that SyMANTIC uncovers similar or more accurate models at a fraction of the cost of existing SR methods.
TorchSISSO: A PyTorch-Based Implementation of the Sure Independence Screening and Sparsifying Operator for Efficient and Interpretable Model Discovery
Oct 02, 2024



Symbolic regression (SR) is a powerful machine learning approach that searches for both the structure and parameters of algebraic models, offering interpretable and compact representations of complex data. Unlike traditional regression methods, SR explores progressively complex feature spaces, which can uncover simple models that generalize well, even from small datasets. Among SR algorithms, the Sure Independence Screening and Sparsifying Operator (SISSO) has proven particularly effective in the natural sciences, helping to rediscover fundamental physical laws as well as discover new interpretable equations for materials property modeling. However, its widespread adoption has been limited by performance inefficiencies and the challenges posed by its FORTRAN-based implementation, especially in modern computing environments. In this work, we introduce TorchSISSO, a native Python implementation built in the PyTorch framework. TorchSISSO leverages GPU acceleration, easy integration, and extensibility, offering a significant speed-up and improved accuracy over the original. We demonstrate that TorchSISSO matches or exceeds the performance of the original SISSO across a range of tasks, while dramatically reducing computational time and improving accessibility for broader scientific applications.
Accelerating Black-Box Molecular Property Optimization by Adaptively Learning Sparse Subspaces
Jan 02, 2024

Molecular property optimization (MPO) problems are inherently challenging since they are formulated over discrete, unstructured spaces and the labeling process involves expensive simulations or experiments, which fundamentally limits the amount of available data. Bayesian optimization (BO) is a powerful and popular framework for efficient optimization of noisy, black-box objective functions (e.g., measured property values), thus is a potentially attractive framework for MPO. To apply BO to MPO problems, one must select a structured molecular representation that enables construction of a probabilistic surrogate model. Many molecular representations have been developed, however, they are all high-dimensional, which introduces important challenges in the BO process -- mainly because the curse of dimensionality makes it difficult to define and perform inference over a suitable class of surrogate models. This challenge has been recently addressed by learning a lower-dimensional encoding of a SMILE or graph representation of a molecule in an unsupervised manner and then performing BO in the encoded space. In this work, we show that such methods have a tendency to "get stuck," which we hypothesize occurs since the mapping from the encoded space to property values is not necessarily well-modeled by a Gaussian process. We argue for an alternative approach that combines numerical molecular descriptors with a sparse axis-aligned Gaussian process model, which is capable of rapidly identifying sparse subspaces that are most relevant to modeling the unknown property function. We demonstrate that our proposed method substantially outperforms existing MPO methods on a variety of benchmark and real-world problems. Specifically, we show that our method can routinely find near-optimal molecules out of a set of more than $>100$k alternatives within 100 or fewer expensive queries.