 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Universal Deep Learning Framework for Materials X-ray Absorption Spectra
Sep 29, 2024X-ray absorption spectroscopy (XAS) is a powerful characterization technique for probing the local chemical environment of absorbing atoms. However, analyzing XAS data presents with significant challenges, often requiring extensive, computationally intensive simulations, as well as significant domain expertise. These limitations hinder the development of fast, robust XAS analysis pipelines that are essential in high-throughput studies and for autonomous experimentation. We address these challenges with a suite of transfer learning approaches for XAS prediction, each uniquely contributing to improved accuracy and efficiency, as demonstrated on K-edge spectra database covering eight 3d transition metals (Ti-Cu). Our framework is built upon three distinct strategies. First, we use M3GNet to derive latent representations of the local chemical environment of absorption sites as input for XAS prediction, achieving up to order-of-magnitude improvements over conventional featurization techniques. Second, we employ a hierarchical transfer learning strategy, training a universal multi-task model across elements before fine-tuning for element-specific predictions. This cascaded approach after element-wise fine-turning yields models that outperform element-specific models by up to 31\%. Third, we implement cross-fidelity transfer learning, adapting a universal model to predict spectra generated by simulation of a different fidelity with a much higher computational cost. This approach improves prediction accuracy by up to 24\% over models trained on the target fidelity alone. Our approach is extendable to XAS prediction for a broader range of elements and offers a generalizable transfer learning framework to enhance other deep-learning models in materials science.
Decoding Structure-Spectrum Relationships with Physically Organized Latent Spaces
Jan 11, 2023

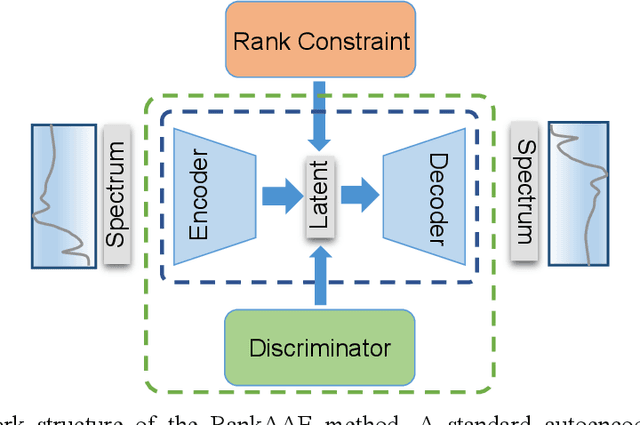

A new semi-supervised machine learning method for the discovery of structure-spectrum relationships is developed and demonstrated using the specific example of interpreting X-ray absorption near-edge structure (XANES) spectra. This method constructs a one-to-one mapping between individual structure descriptors and spectral trends. Specifically, an adversarial autoencoder is augmented with a novel rank constraint (RankAAE). The RankAAE methodology produces a continuous and interpretable latent space, where each dimension can track an individual structure descriptor. As a part of this process, the model provides a robust and quantitative measure of the structure-spectrum relationship by decoupling intertwined spectral contributions from multiple structural characteristics. This makes it ideal for spectral interpretation and the discovery of new descriptors. The capability of this procedure is showcased by considering five local structure descriptors and a database of over fifty thousand simulated XANES spectra across eight first-row transition metal oxide families. The resulting structure-spectrum relationships not only reproduce known trends in the literature, but also reveal unintuitive ones that are visually indiscernible in large data sets. The results suggest that the RankAAE methodology has great potential to assist researchers to interpret complex scientific data, test physical hypotheses, and reveal new patterns that extend scientific insight.