 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBASiNETEntropy: an alignment-free method for classification of biological sequences through complex networks and entropy maximization
Mar 24, 2022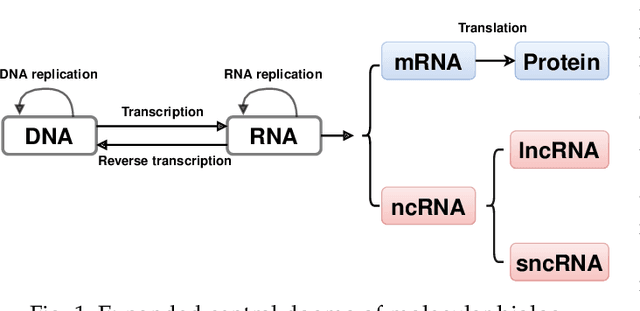

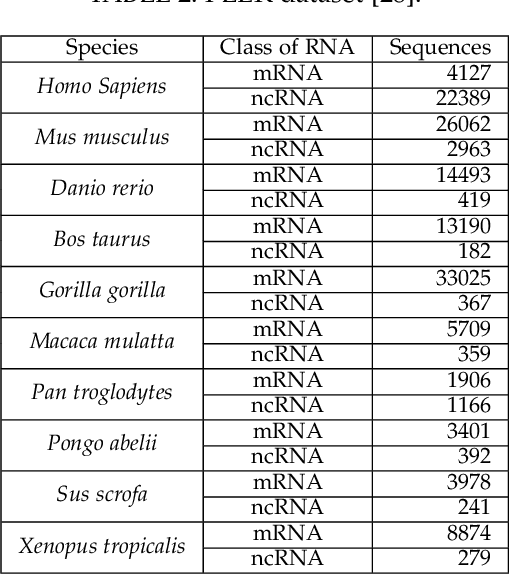
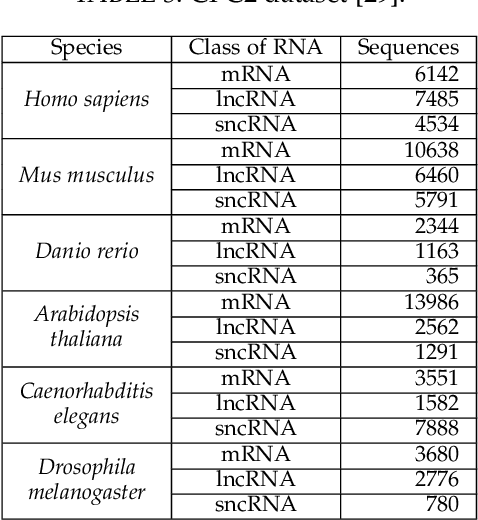
The discovery of nucleic acids and the structure of DNA have brought considerable advances in the understanding of life. The development of next-generation sequencing technologies has led to a large-scale generation of data, for which computational methods have become essential for analysis and knowledge discovery. In particular, RNAs have received much attention because of the diversity of their functionalities in the organism and the discoveries of different classes with different functions in many biological processes. Therefore, the correct identification of RNA sequences is increasingly important to provide relevant information to understand the functioning of organisms. This work addresses this context by presenting a new method for the classification of biological sequences through complex networks and entropy maximization. The maximum entropy principle is proposed to identify the most informative edges about the RNA class, generating a filtered complex network. The proposed method was evaluated in the classification of different RNA classes from 13 species. The proposed method was compared to PLEK, CPC2 and BASiNET methods, outperforming all compared methods. BASiNETEntropy classified all RNA sequences with high accuracy and low standard deviation in results, showing assertiveness and robustness. The proposed method is implemented in an open source in R language and is freely available at https://cran.r-project.org/web/packages/BASiNETEntropy.
Complex Network-Based Approach for Feature Extraction and Classification of Musical Genres
Oct 09, 2021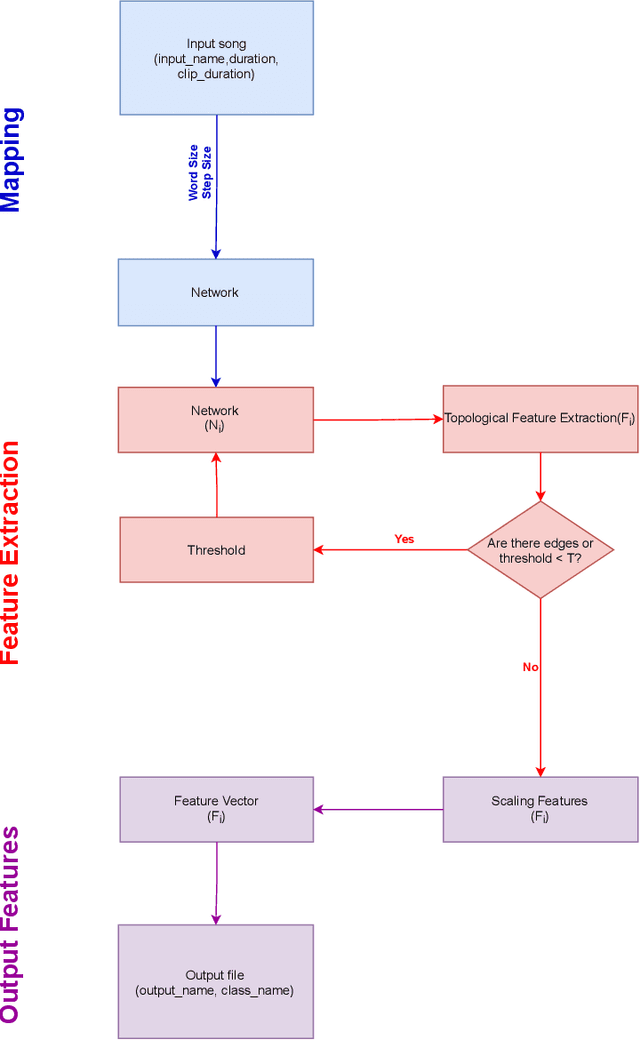
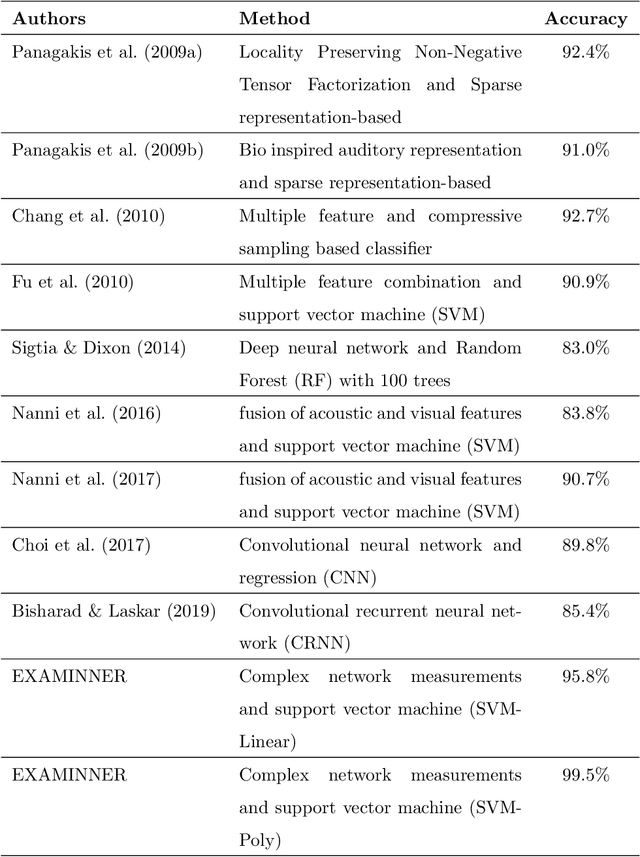
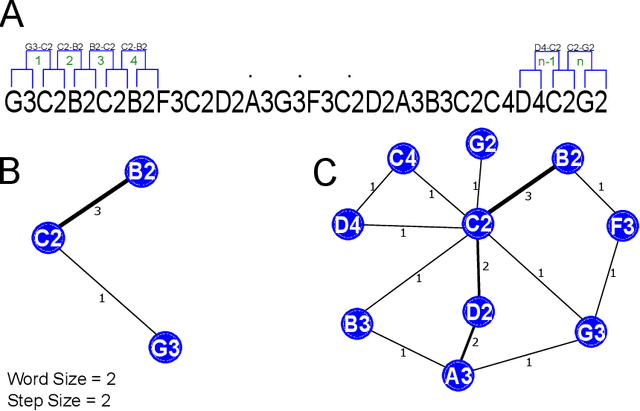
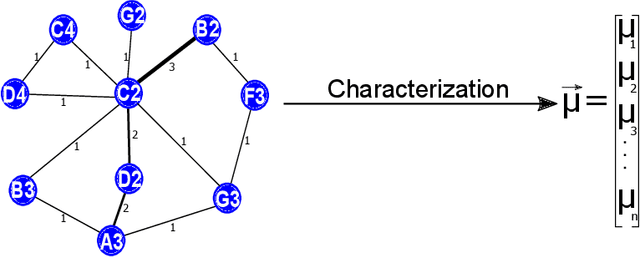
Musical genre's classification has been a relevant research topic. The association between music and genres is fundamental for the media industry, which manages musical recommendation systems, and for music streaming services, which may appear classified by genres. In this context, this work presents a feature extraction method for the automatic classification of musical genres, based on complex networks and their topological measurements. The proposed method initially converts the musics into sequences of musical notes and then maps the sequences as complex networks. Topological measurements are extracted to characterize the network topology, which composes a feature vector that applies to the classification of musical genres. The method was evaluated in the classification of 10 musical genres by adopting the GTZAN dataset and 8 musical genres by adopting the FMA dataset. The results were compared with methods in the literature. The proposed method outperformed all compared methods by presenting high accuracy and low standard deviation, showing its suitability for the musical genre's classification, which contributes to the media industry in the automatic classification with assertiveness and robustness. The proposed method is implemented in an open source in the Python language and freely available at https://github.com/omatheuspimenta/examinner.
Feature extraction from complex networks: A case of study in genomic sequences classification
Dec 17, 2014

This work presents a new approach for classification of genomic sequences from measurements of complex networks and information theory. For this, it is considered the nucleotides, dinucleotides and trinucleotides of a genomic sequence. For each of them, the entropy, sum entropy and maximum entropy values are calculated.For each of them is also generated a network, in which the nodes are the nucleotides, dinucleotides or trinucleotides and its edges are estimated by observing the respective adjacency among them in the genomic sequence. In this way, it is generated three networks, for which measures of complex networks are extracted.These measures together with measures of information theory comprise a feature vector representing a genomic sequence. Thus, the feature vector is used for classification by methods such as SVM, MultiLayer Perceptron, J48, IBK, Naive Bayes and Random Forest in order to evaluate the proposed approach.It was adopted coding sequences, intergenic sequences and TSS (Transcriptional Starter Sites) as datasets, for which the better results were obtained by the Random Forest with 91.2%, followed by J48 with 89.1% and SVM with 84.8% of accuracy. These results indicate that the new approach of feature extraction has its value, reaching good levels of classification even considering only the genomic sequences, i.e., no other a priori knowledge about them is considered.
An iterative feature selection method for GRNs inference by exploring topological properties
Jul 25, 2011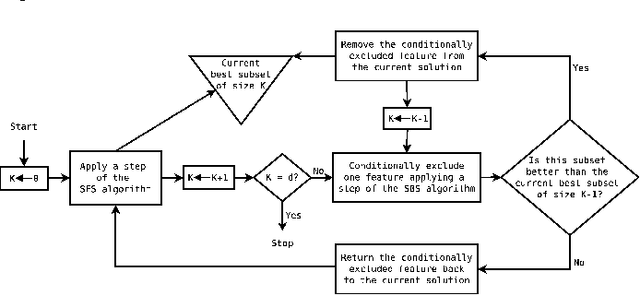
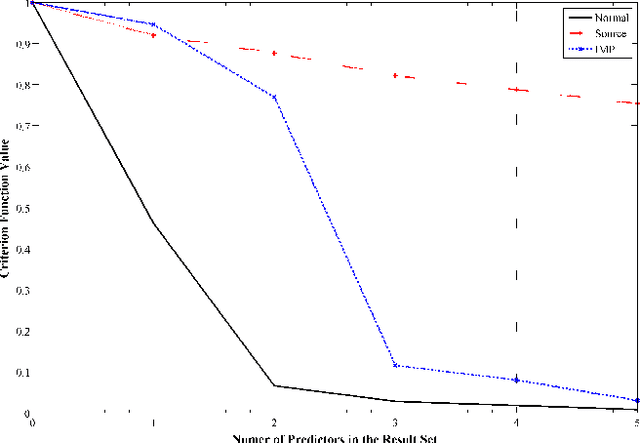
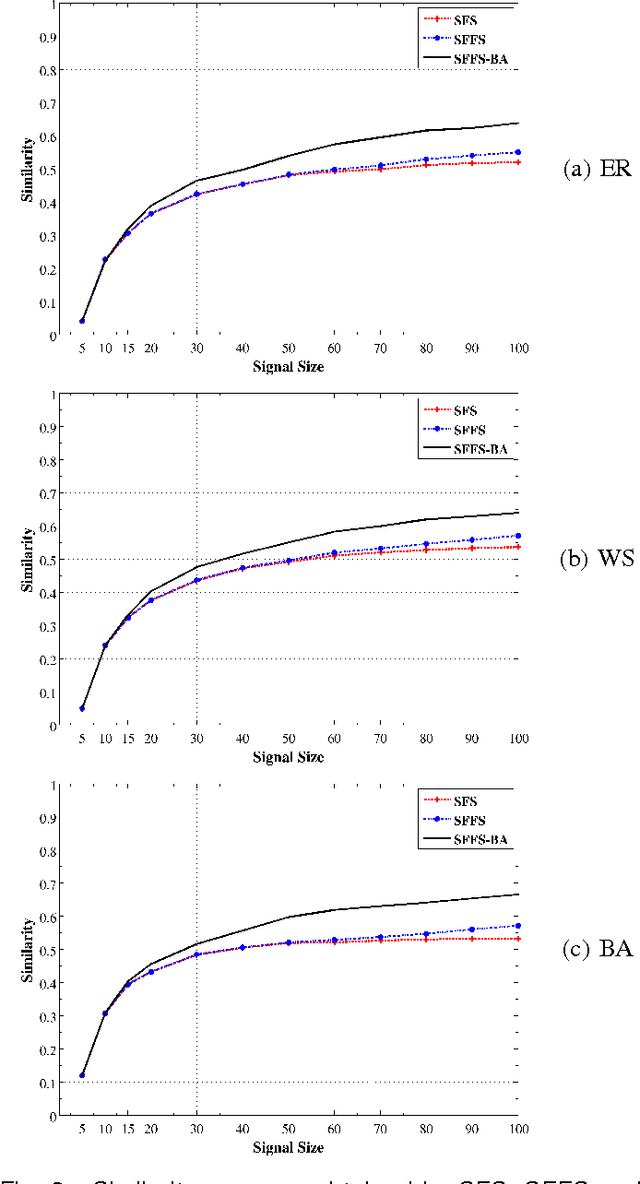
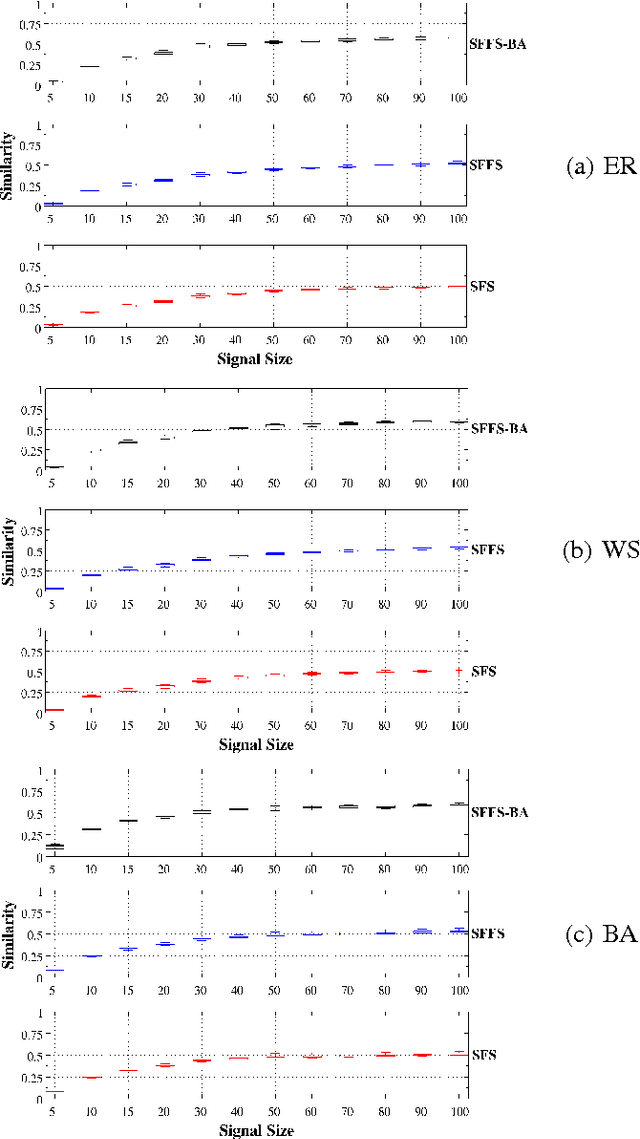
An important problem in bioinformatics is the inference of gene regulatory networks (GRN) from temporal expression profiles. In general, the main limitations faced by GRN inference methods is the small number of samples with huge dimensionalities and the noisy nature of the expression measurements. In face of these limitations, alternatives are needed to get better accuracy on the GRNs inference problem. This work addresses this problem by presenting an alternative feature selection method that applies prior knowledge on its search strategy, called SFFS-BA. The proposed search strategy is based on the Sequential Floating Forward Selection (SFFS) algorithm, with the inclusion of a scale-free (Barab\'asi-Albert) topology information in order to guide the search process to improve inference. The proposed algorithm explores the scale-free property by pruning the search space and using a power law as a weight for reducing it. In this way, the search space traversed by the SFFS-BA method combines a breadth-first search when the number of combinations is small (<k> <= 2) with a depth-first search when the number of combinations becomes explosive (<k> >= 3), being guided by the scale-free prior information. Experimental results show that the SFFS-BA provides a better inference similarities than SFS and SFFS, keeping the robustness of the SFS and SFFS methods, thus presenting very good results.