 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Investigation of Hepatitis B Virus Genome using Markov Models
Nov 12, 2023
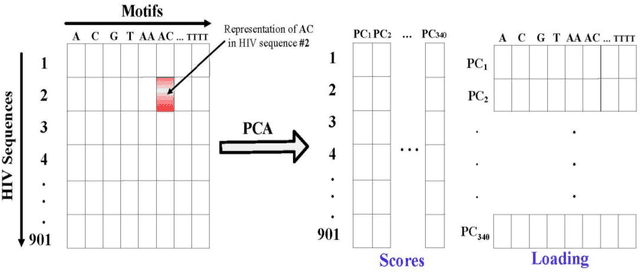
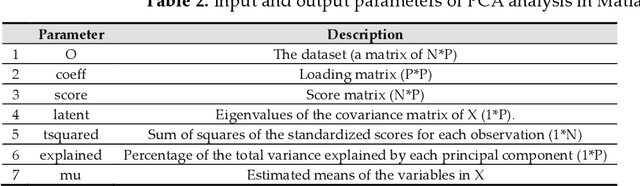
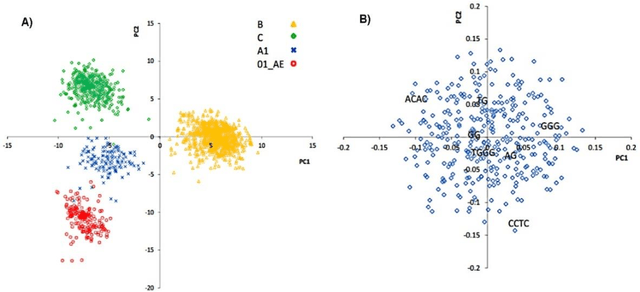
The human genome encodes a family of editing enzymes known as APOBEC3 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3). Several family members, such as APO-BEC3G, APOBEC3F, and APOBEC3H haplotype II, exhibit activity against viruses such as HIV. These enzymes induce C-to-U mutations in the negative strand of viral genomes, resulting in multiple G-to-A changes, commonly referred to as 'hypermutation.' Mutations catalyzed by these enzymes are sequence context-dependent in the HIV genome; for instance, APOBEC3G preferen-tially mutates G within GG, TGG, and TGGG contexts, while other members mutate G within GA, TGA, and TGAA contexts. However, the same sequence context has not been explored in relation to these enzymes and HBV. In this study, our objective is to identify the mutational footprint of APOBEC3 enzymes in the HBV genome. To achieve this, we employ a multivariable data analytics technique to investigate motif preferences and potential sequence hierarchies of mutation by APOBEC3 enzymes using full genome HBV sequences from a diverse range of naturally infected patients. This approach allows us to distinguish between normal and hypermutated sequences based on the representation of mono- to tetra-nucleotide motifs. Additionally, we aim to identify motifs associated with hypermutation induced by different APOBEC3 enzymes in HBV genomes. Our analyses reveal that either APOBEC3 enzymes are not active against HBV, or the induction of G-to-A mutations by these enzymes is not sequence context-dependent in the HBV genome.