 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnsuring Responsible Sourcing of Large Language Model Training Data Through Knowledge Graph Comparison
Jul 02, 2024In light of recent plagiarism allegations Brough by publishers, newspapers, and other creators of copyrighted corpora against large language model (LLM) developers, we propose a novel system, a variant of a plagiarism detection system, that assesses whether a knowledge source has been used in the training or fine-tuning of a large language model. Unlike current methods, we utilize an approach that uses Resource Description Framework (RDF) triples to create knowledge graphs from both a source document and a LLM continuation of that document. These graphs are then analyzed with respect to content using cosine similarity and with respect to structure using a normalized version of graph edit distance that shows the degree of isomorphism. Unlike traditional systems that focus on content matching and keyword identification between a source and target corpus, our approach enables a broader evaluation of similarity and thus a more accurate comparison of the similarity between a source document and LLM continuation by focusing on relationships between ideas and their organization with regards to others. Additionally, our approach does not require access to LLM metrics like perplexity that may be unavailable in closed large language modeling "black-box" systems, as well as the training corpus. A prototype of our system will be found on a hyperlinked GitHub repository.
SeqMate: A Novel Large Language Model Pipeline for Automating RNA Sequencing
Jul 02, 2024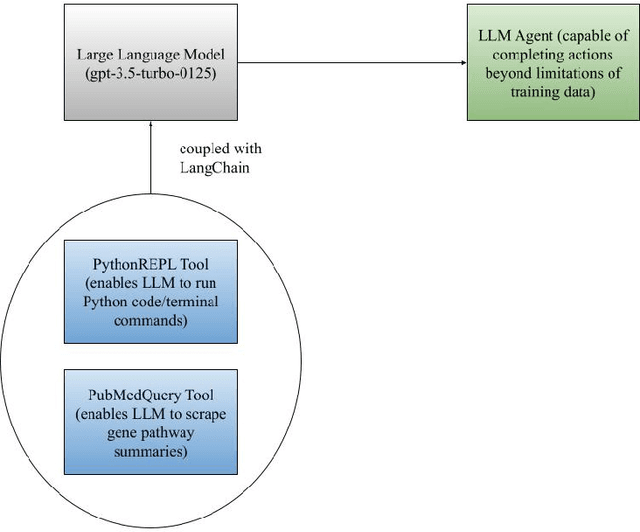
RNA sequencing techniques, like bulk RNA-seq and Single Cell (sc) RNA-seq, are critical tools for the biologist looking to analyze the genetic activity/transcriptome of a tissue or cell during an experimental procedure. Platforms like Illumina's next-generation sequencing (NGS) are used to produce the raw data for this experimental procedure. This raw FASTQ data must then be prepared via a complex series of data manipulations by bioinformaticians. This process currently takes place on an unwieldy textual user interface like a terminal/command line that requires the user to install and import multiple program packages, preventing the untrained biologist from initiating data analysis. Open-source platforms like Galaxy have produced a more user-friendly pipeline, yet the visual interface remains cluttered and highly technical, remaining uninviting for the natural scientist. To address this, SeqMate is a user-friendly tool that allows for one-click analytics by utilizing the power of a large language model (LLM) to automate both data preparation and analysis (differential expression, trajectory analysis, etc). Furthermore, by utilizing the power of generative AI, SeqMate is also capable of analyzing such findings and producing written reports of upregulated/downregulated/user-prompted genes with sources cited from known repositories like PubMed, PDB, and Uniprot.
Reducing Large Language Model Bias with Emphasis on 'Restricted Industries': Automated Dataset Augmentation and Prejudice Quantification
Mar 20, 2024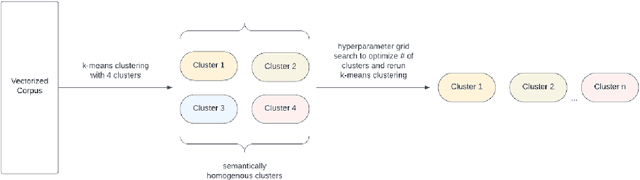
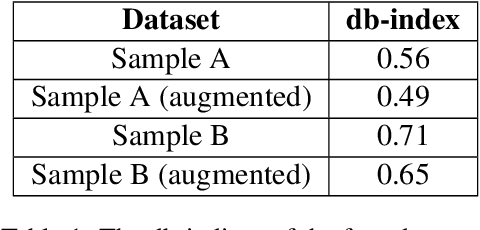
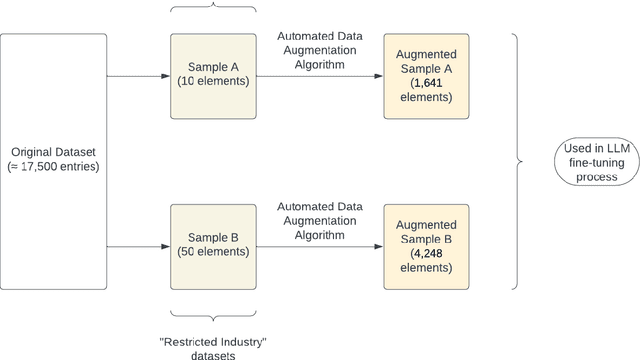
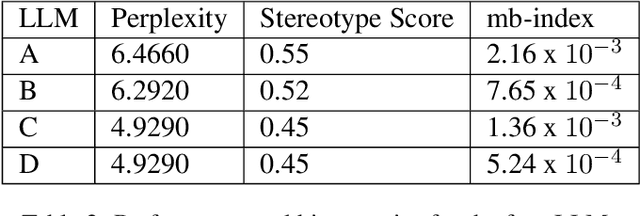
Despite the growing capabilities of large language models, there exists concerns about the biases they develop. In this paper, we propose a novel, automated mechanism for debiasing through specified dataset augmentation in the lens of bias producers and in the context of 'restricted industries' with limited data. We additionally create two new additional metrics, the mb-index and db-index, to quantify bias, considering the idea that bias occurs due to both intrinsic model architecture and dataset.