 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpretable Deep Learning for Polar Mechanistic Reaction Prediction
Apr 22, 2025Accurately predicting chemical reactions is essential for driving innovation in synthetic chemistry, with broad applications in medicine, manufacturing, and agriculture. At the same time, reaction prediction is a complex problem which can be both time-consuming and resource-intensive for chemists to solve. Deep learning methods offer an appealing solution by enabling high-throughput reaction prediction. However, many existing models are trained on the US Patent Office dataset and treat reactions as overall transformations: mapping reactants directly to products with limited interpretability or mechanistic insight. To address this, we introduce PMechRP (Polar Mechanistic Reaction Predictor), a system that trains machine learning models on the PMechDB dataset, which represents reactions as polar elementary steps that capture electron flow and mechanistic detail. To further expand model coverage and improve generalization, we augment PMechDB with a diverse set of combinatorially generated reactions. We train and compare a range of machine learning models, including transformer-based, graph-based, and two-step siamese architectures. Our best-performing approach was a hybrid model, which combines a 5-ensemble of Chemformer models with a two-step Siamese framework to leverage the accuracy of transformer architectures, while filtering away "alchemical" products using the two-step network predictions. For evaluation, we use a test split of the PMechDB dataset and additionally curate a human benchmark dataset consisting of complete mechanistic pathways extracted from an organic chemistry textbook. Our hybrid model achieves a top-10 accuracy of 94.9% on the PMechDB test set and a target recovery rate of 84.9% on the pathway dataset.
AI for Interpretable Chemistry: Predicting Radical Mechanistic Pathways via Contrastive Learning
Nov 02, 2023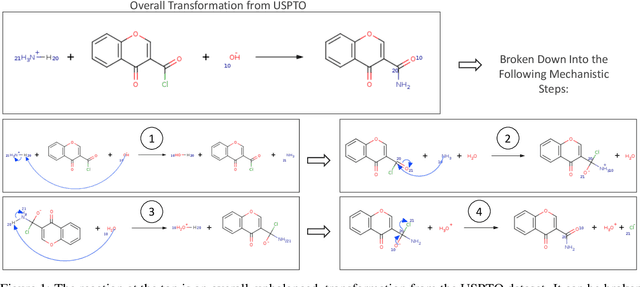
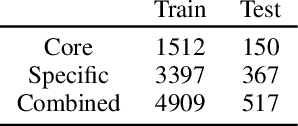
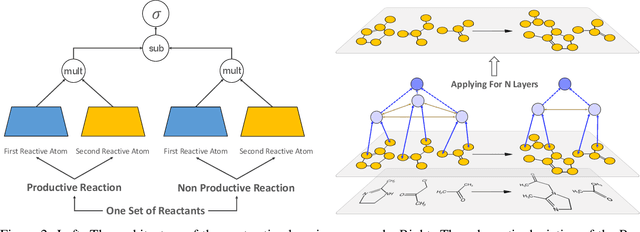
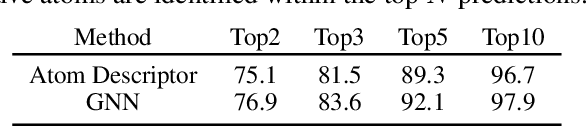
Deep learning-based reaction predictors have undergone significant architectural evolution. However, their reliance on reactions from the US Patent Office results in a lack of interpretable predictions and limited generalization capability to other chemistry domains, such as radical and atmospheric chemistry. To address these challenges, we introduce a new reaction predictor system, RMechRP, that leverages contrastive learning in conjunction with mechanistic pathways, the most interpretable representation of chemical reactions. Specifically designed for radical reactions, RMechRP provides different levels of interpretation of chemical reactions. We develop and train multiple deep-learning models using RMechDB, a public database of radical reactions, to establish the first benchmark for predicting radical reactions. Our results demonstrate the effectiveness of RMechRP in providing accurate and interpretable predictions of radical reactions, and its potential for various applications in atmospheric chemistry.
Quantum Mechanics and Machine Learning Synergies: Graph Attention Neural Networks to Predict Chemical Reactivity
Mar 24, 2021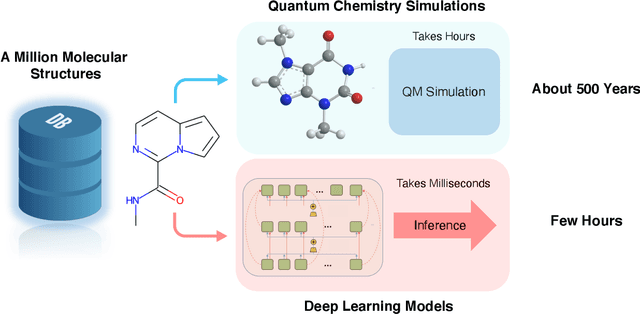
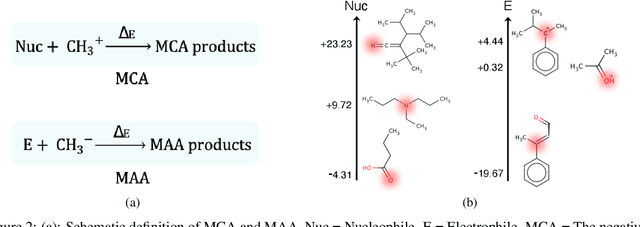
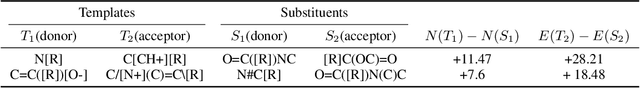
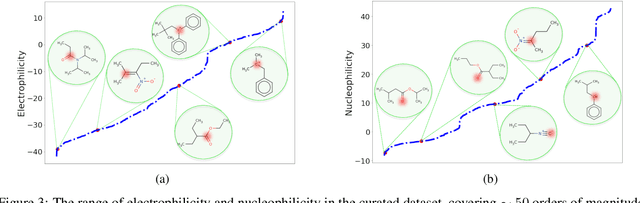
There is a lack of scalable quantitative measures of reactivity for functional groups in organic chemistry. Measuring reactivity experimentally is costly and time-consuming and does not scale to the astronomical size of chemical space. In previous quantum chemistry studies, we have introduced Methyl Cation Affinities (MCA*) and Methyl Anion Affinities (MAA*), using a solvation model, as quantitative measures of reactivity for organic functional groups over the broadest range. Although MCA* and MAA* offer good estimates of reactivity parameters, their calculation through Density Functional Theory (DFT) simulations is time-consuming. To circumvent this problem, we first use DFT to calculate MCA* and MAA* for more than 2,400 organic molecules thereby establishing a large dataset of chemical reactivity scores. We then design deep learning methods to predict the reactivity of molecular structures and train them using this curated dataset in combination with different representations of molecular structures. Using ten-fold cross-validation, we show that graph attention neural networks applied to informative input fingerprints produce the most accurate estimates of reactivity, achieving over 91% test accuracy for predicting the MCA* plus-minus 3.0 or MAA* plus-minus 3.0, over 50 orders of magnitude. Finally, we demonstrate the application of these reactivity scores to two tasks: (1) chemical reaction prediction; (2) combinatorial generation of reaction mechanisms. The curated dataset of MCA* and MAA* scores is available through the ChemDB chemoinformatics web portal at www.cdb.ics.uci.edu.