 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQuantum Mechanics and Machine Learning Synergies: Graph Attention Neural Networks to Predict Chemical Reactivity
Paper and Code
Mar 24, 2021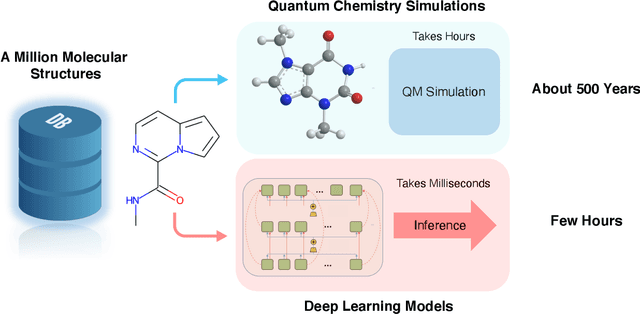
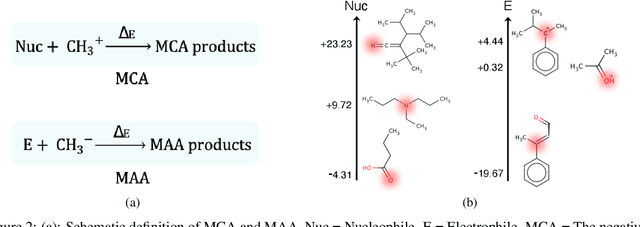
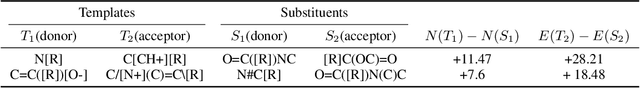
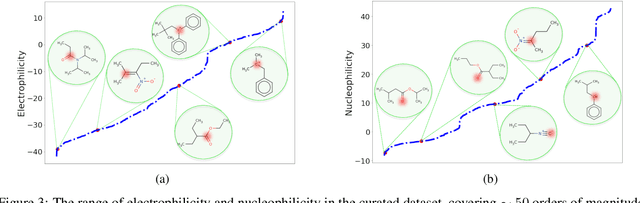
There is a lack of scalable quantitative measures of reactivity for functional groups in organic chemistry. Measuring reactivity experimentally is costly and time-consuming and does not scale to the astronomical size of chemical space. In previous quantum chemistry studies, we have introduced Methyl Cation Affinities (MCA*) and Methyl Anion Affinities (MAA*), using a solvation model, as quantitative measures of reactivity for organic functional groups over the broadest range. Although MCA* and MAA* offer good estimates of reactivity parameters, their calculation through Density Functional Theory (DFT) simulations is time-consuming. To circumvent this problem, we first use DFT to calculate MCA* and MAA* for more than 2,400 organic molecules thereby establishing a large dataset of chemical reactivity scores. We then design deep learning methods to predict the reactivity of molecular structures and train them using this curated dataset in combination with different representations of molecular structures. Using ten-fold cross-validation, we show that graph attention neural networks applied to informative input fingerprints produce the most accurate estimates of reactivity, achieving over 91% test accuracy for predicting the MCA* plus-minus 3.0 or MAA* plus-minus 3.0, over 50 orders of magnitude. Finally, we demonstrate the application of these reactivity scores to two tasks: (1) chemical reaction prediction; (2) combinatorial generation of reaction mechanisms. The curated dataset of MCA* and MAA* scores is available through the ChemDB chemoinformatics web portal at www.cdb.ics.uci.edu.