 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProgramming and Training Rate-Independent Chemical Reaction Networks
Sep 20, 2021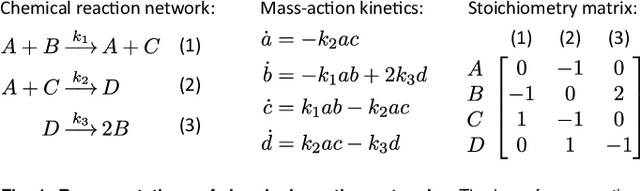
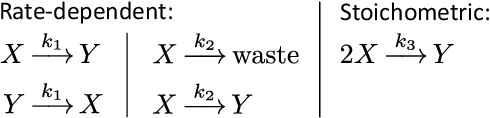
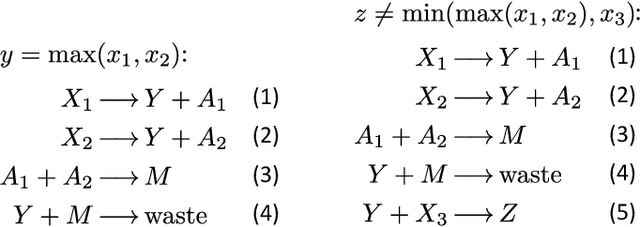
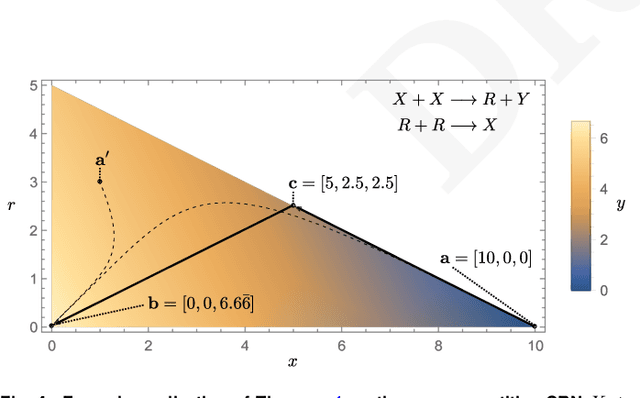
Embedding computation in biochemical environments incompatible with traditional electronics is expected to have wide-ranging impact in synthetic biology, medicine, nanofabrication and other fields. Natural biochemical systems are typically modeled by chemical reaction networks (CRNs), and CRNs can be used as a specification language for synthetic chemical computation. In this paper, we identify a class of CRNs called non-competitive (NC) whose equilibria are absolutely robust to reaction rates and kinetic rate law, because their behavior is captured solely by their stoichiometric structure. Unlike prior work on rate-independent CRNs, checking non-competition and using it as a design criterion is easy and promises robust output. We also present a technique to program NC-CRNs using well-founded deep learning methods, showing a translation procedure from rectified linear unit (ReLU) neural networks to NC-CRNs. In the case of binary weight ReLU networks, our translation procedure is surprisingly tight in the sense that a single bimolecular reaction corresponds to a single ReLU node and vice versa. This compactness argues that neural networks may be a fitting paradigm for programming rate-independent chemical computation. As proof of principle, we demonstrate our scheme with numerical simulations of CRNs translated from neural networks trained on traditional machine learning datasets (IRIS and MNIST), as well as tasks better aligned with potential biological applications including virus detection and spatial pattern formation.
Deep Molecular Programming: A Natural Implementation of Binary-Weight ReLU Neural Networks
Apr 21, 2020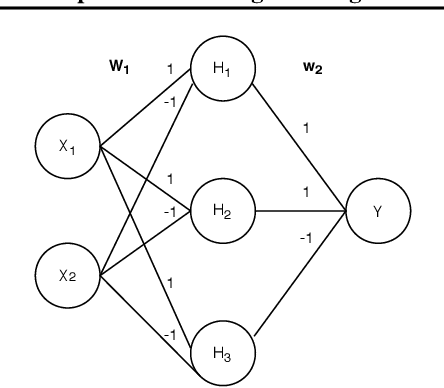
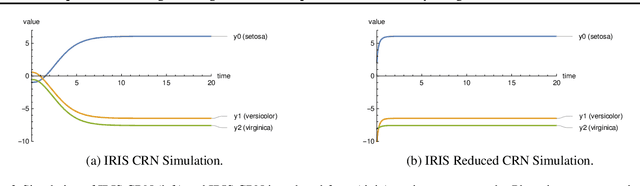
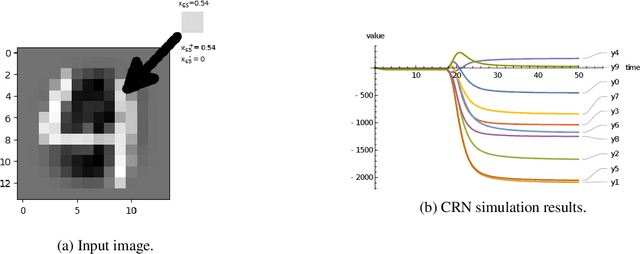
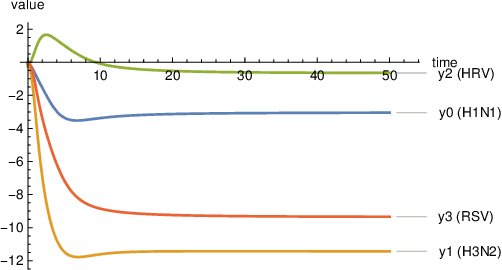
Embedding computation in molecular contexts incompatible with traditional electronics is expected to have wide ranging impact in synthetic biology, medicine, nanofabrication and other fields. A key remaining challenge lies in developing programming paradigms for molecular computation that are well-aligned with the underlying chemical hardware and do not attempt to shoehorn ill-fitting electronics paradigms. We discover a surprisingly tight connection between a popular class of neural networks (Binary-weight ReLU aka BinaryConnect) and a class of coupled chemical reactions that are absolutely robust to reaction rates. The robustness of rate-independent chemical computation makes it a promising target for bioengineering implementation. We show how a BinaryConnect neural network trained in silico using well-founded deep learning optimization techniques, can be compiled to an equivalent chemical reaction network, providing a novel molecular programming paradigm. We illustrate such translation on the paradigmatic IRIS and MNIST datasets. Toward intended applications of chemical computation, we further use our method to generate a CRN that can discriminate between different virus types based on gene expression levels. Our work sets the stage for rich knowledge transfer between neural network and molecular programming communities.
Statistical Learning of Arbitrary Computable Classifiers
Jul 10, 2008Statistical learning theory chiefly studies restricted hypothesis classes, particularly those with finite Vapnik-Chervonenkis (VC) dimension. The fundamental quantity of interest is the sample complexity: the number of samples required to learn to a specified level of accuracy. Here we consider learning over the set of all computable labeling functions. Since the VC-dimension is infinite and a priori (uniform) bounds on the number of samples are impossible, we let the learning algorithm decide when it has seen sufficient samples to have learned. We first show that learning in this setting is indeed possible, and develop a learning algorithm. We then show, however, that bounding sample complexity independently of the distribution is impossible. Notably, this impossibility is entirely due to the requirement that the learning algorithm be computable, and not due to the statistical nature of the problem.