 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePlasmoFAB: A Benchmark to Foster Machine Learning for Plasmodium falciparum Protein Antigen Candidate Prediction
Jan 16, 2023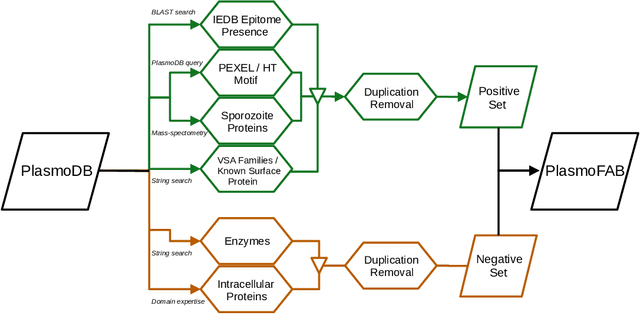
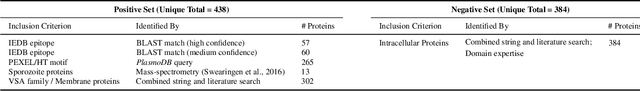
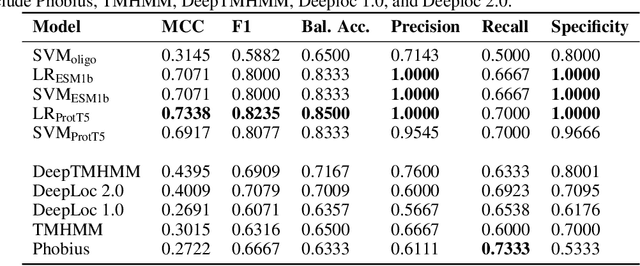
Motivation: Machine learning methods can be used to support scientific discovery in healthcare-related research fields. However, these methods can only be reliably used if they can be trained on high-quality and curated datasets. Currently, no such dataset for the exploration of Plasmodium falciparum protein antigen candidates exists. The parasite Plasmodium falciparum causes the infectious disease malaria. Thus, identifying potential antigens is of utmost importance for the development of antimalarial drugs and vaccines. Since exploring antigen candidates experimentally is an expensive and time-consuming process, applying machine learning methods to support this process has the potential to accelerate the development of drugs and vaccines which are needed for fighting and controlling malaria. Results: We developed PlasmoFAB, a curated benchmark that can be used to train machine learning methods for the exploration of Plasmodium falciparum protein antigen candidates. We combined an extensive literature search with domain expertise to create high-quality labels for Plasmodium falciparum specific proteins that distinguish between antigen candidates and intracellular proteins. Additionally, we used our benchmark to compare different well-known prediction models and available protein localization prediction services on the task of identifying protein antigen candidates. We show that available general-purpose services are unable to provide sufficient performance on identifying protein antigen candidates and are outperformed by models that were trained on specialized data.
COmic: Convolutional Kernel Networks for Interpretable End-to-End Learning on (Multi-)Omics Data
Dec 02, 2022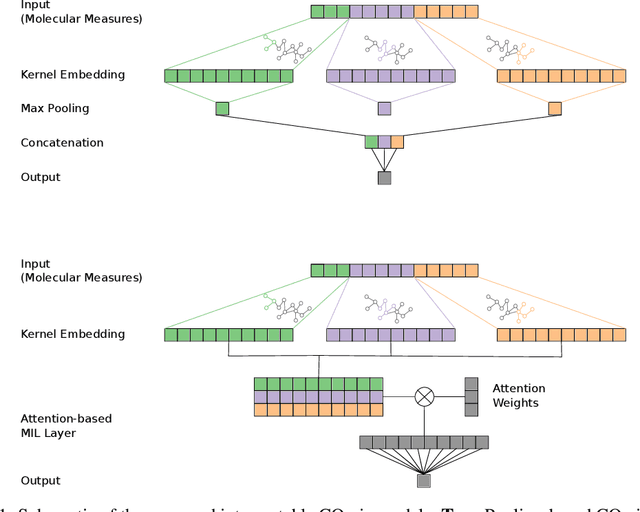
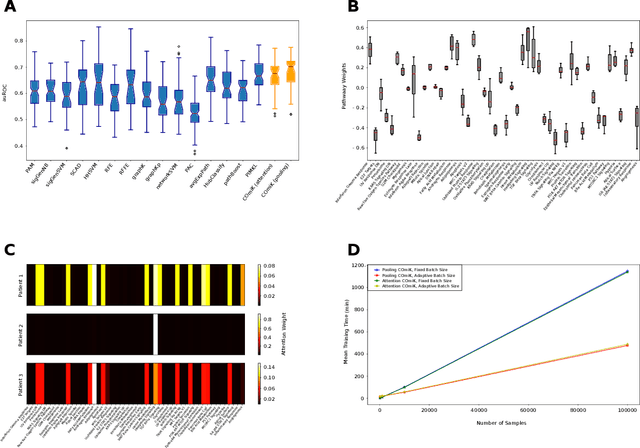
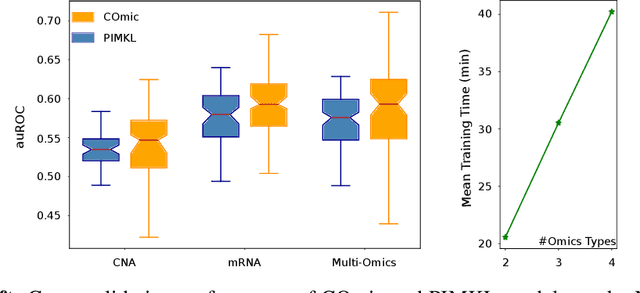
Motivation: The size of available omics datasets is steadily increasing with technological advancement in recent years. While this increase in sample size can be used to improve the performance of relevant prediction tasks in healthcare, models that are optimized for large datasets usually operate as black boxes. In high stakes scenarios, like healthcare, using a black-box model poses safety and security issues. Without an explanation about molecular factors and phenotypes that affected the prediction, healthcare providers are left with no choice but to blindly trust the models. We propose a new type of artificial neural networks, named Convolutional Omics Kernel Networks (COmic). By combining convolutional kernel networks with pathway-induced kernels, our method enables robust and interpretable end-to-end learning on omics datasets ranging in size from a few hundred to several hundreds of thousands of samples. Furthermore, COmic can be easily adapted to utilize multi-omics data. Results: We evaluate the performance capabilities of COmic on six different breast cancer cohorts. Additionally, we train COmic models on multi-omics data using the METABRIC cohort. Our models perform either better or similar to competitors on both tasks. We show how the use of pathway-induced Laplacian kernels opens the black-box nature of neural networks and results in intrinsically interpretable models that eliminate the need for \textit{post-hoc} explanation models.
Convolutional Motif Kernel Networks
Nov 03, 2021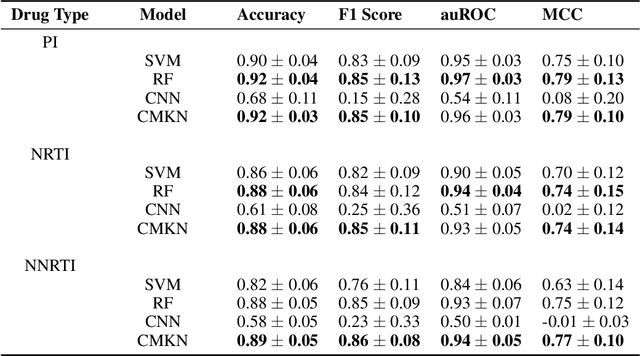
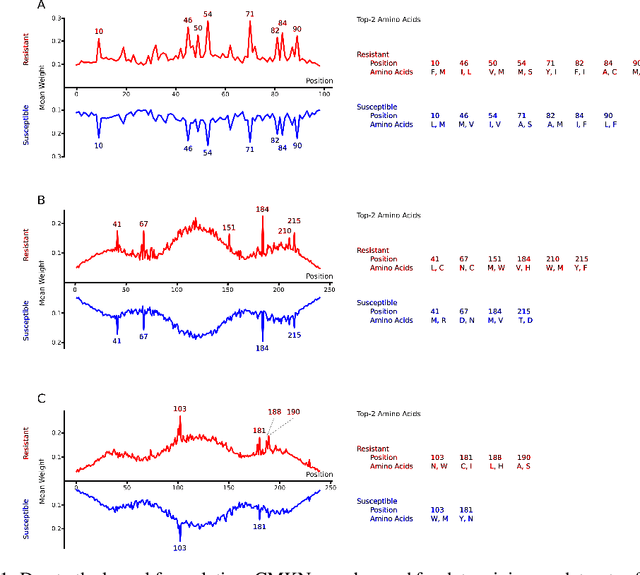
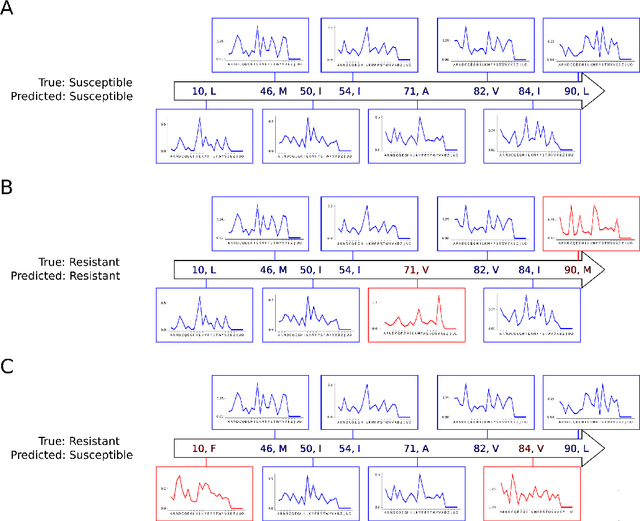
Artificial neural networks are exceptionally good in learning to detect correlations within data that are associated with specified outcomes. However to deepen knowledge and support further research, researchers have to be able to explain predicted outcomes within the data's domain. Furthermore, domain experts like Healthcare Providers need these explanations to assess whether a predicted outcome can be trusted in high stakes scenarios and to help them incorporating a model into their own routine. In this paper we introduce Convolutional Motif Kernel Networks, a neural network architecture that incorporates learning a feature representation within a subspace of the reproducing kernel Hilbert space of the motif kernel function. The resulting model has state-of-the-art performance and enables researchers and domain experts to directly interpret and verify prediction outcomes without the need for a post hoc explainability method.