 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSAERMA: Stacked Autoencoder Rule Mining Algorithm for the Interpretation of Epistatic Interactions in GWAS for Extreme Obesity
Aug 27, 2019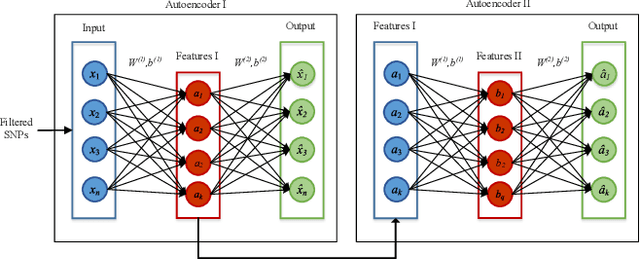
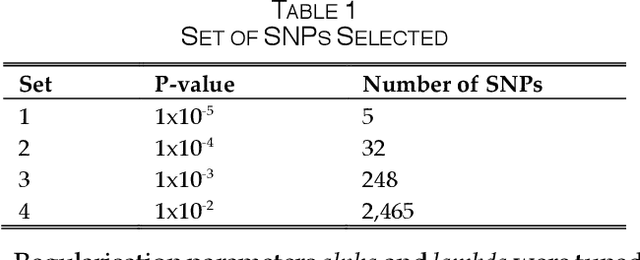
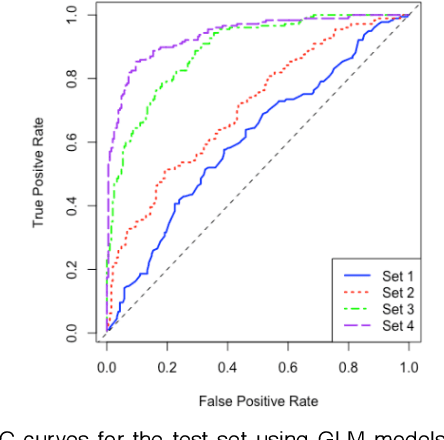
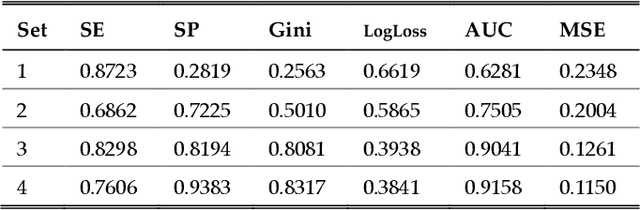
One of the most important challenges in the analysis of high-throughput genetic data is the development of efficient computational methods to identify statistically significant Single Nucleotide Polymorphisms (SNPs). Genome-wide association studies (GWAS) use single-locus analysis where each SNP is independently tested for association with phenotypes. The limitation with this approach, however, is its inability to explain genetic variation in complex diseases. Alternative approaches are required to model the intricate relationships between SNPs. Our proposed approach extends GWAS by combining deep learning stacked autoencoders (SAEs) and association rule mining (ARM) to identify epistatic interactions between SNPs. Following traditional GWAS quality control and association analysis, the most significant SNPs are selected and used in the subsequent analysis to investigate epistasis. SAERMA controls the classification results produced in the final fully connected multi-layer feedforward artificial neural network (MLP) by manipulating the interestingness measures, support and confidence, in the rule generation process. The best classification results were achieved with 204 SNPs compressed to 100 units (77% AUC, 77% SE, 68% SP, 53% Gini, logloss=0.58, and MSE=0.20), although it was possible to achieve 73% AUC (77% SE, 63% SP, 45% Gini, logloss=0.62, and MSE=0.21) with 50 hidden units - both supported by close model interpretation.
Extracting Epistatic Interactions in Type 2 Diabetes Genome-Wide Data Using Stacked Autoencoder
Aug 28, 2018
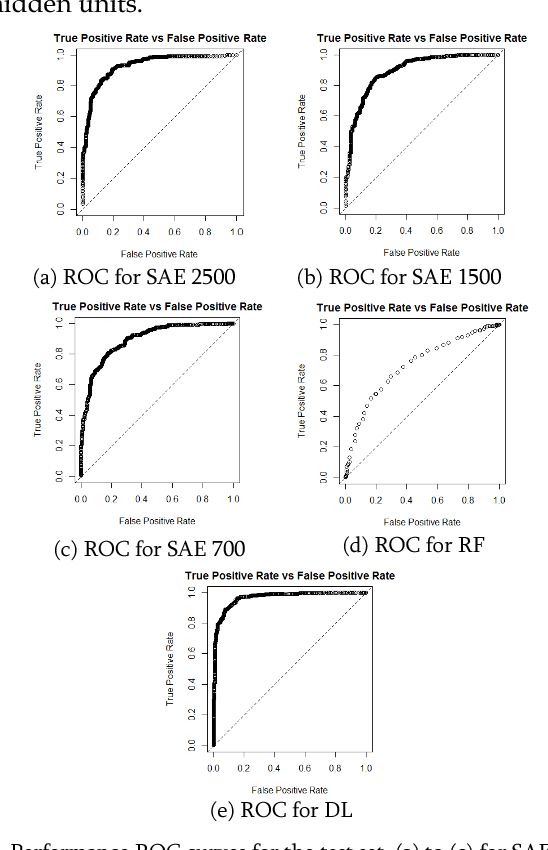
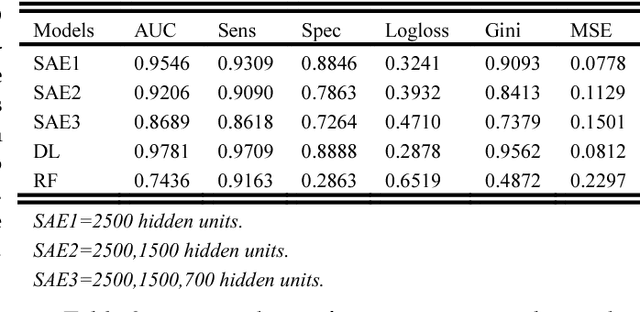
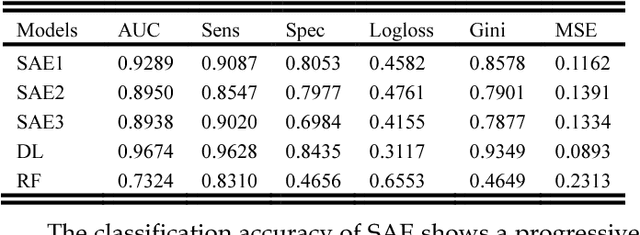
2 Diabetes is a leading worldwide public health concern, and its increasing prevalence has significant health and economic importance in all nations. The condition is a multifactorial disorder with a complex aetiology. The genetic determinants remain largely elusive, with only a handful of identified candidate genes. Genome wide association studies (GWAS) promised to significantly enhance our understanding of genetic based determinants of common complex diseases. To date, 83 single nucleotide polymorphisms (SNPs) for type 2 diabetes have been identified using GWAS. Standard statistical tests for single and multi-locus analysis such as logistic regression, have demonstrated little effect in understanding the genetic architecture of complex human diseases. Logistic regression is modelled to capture linear interactions but neglects the non-linear epistatic interactions present within genetic data. There is an urgent need to detect epistatic interactions in complex diseases as this may explain the remaining missing heritability in such diseases. In this paper, we present a novel framework based on deep learning algorithms that deal with non-linear epistatic interactions that exist in genome wide association data. Logistic association analysis under an additive genetic model, adjusted for genomic control inflation factor, is conducted to remove statistically improbable SNPs to minimize computational overheads.