 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCryoSAMU: Enhancing 3D Cryo-EM Density Maps of Protein Structures at Intermediate Resolution with Structure-Aware Multimodal U-Nets
Mar 26, 2025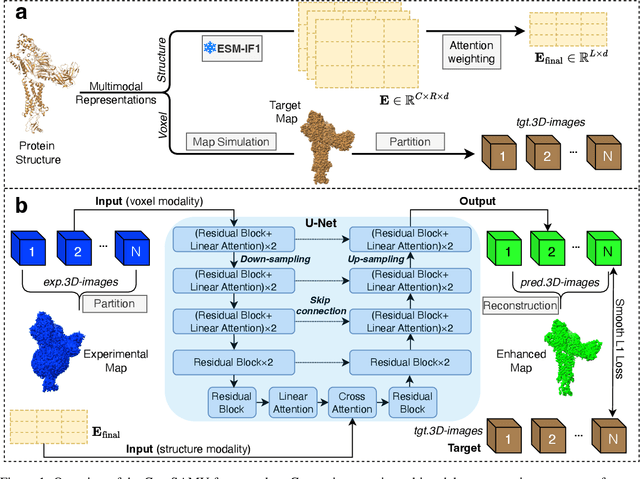
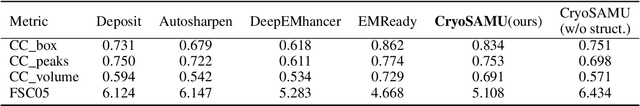

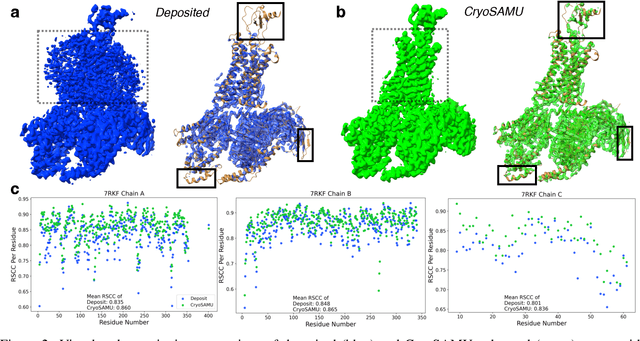
Enhancing cryogenic electron microscopy (cryo-EM) 3D density maps at intermediate resolution (4-8 {\AA}) is crucial in protein structure determination. Recent advances in deep learning have led to the development of automated approaches for enhancing experimental cryo-EM density maps. Yet, these methods are not optimized for intermediate-resolution maps and rely on map density features alone. To address this, we propose CryoSAMU, a novel method designed to enhance 3D cryo-EM density maps of protein structures using structure-aware multimodal U-Nets and trained on curated intermediate-resolution density maps. We comprehensively evaluate CryoSAMU across various metrics and demonstrate its competitive performance compared to state-of-the-art methods. Notably, CryoSAMU achieves significantly faster processing speed, showing promise for future practical applications. Our code is available at https://github.com/chenwei-zhang/CryoSAMU.
Synthetic High-resolution Cryo-EM Density Maps with Generative Adversarial Networks
Jul 24, 2024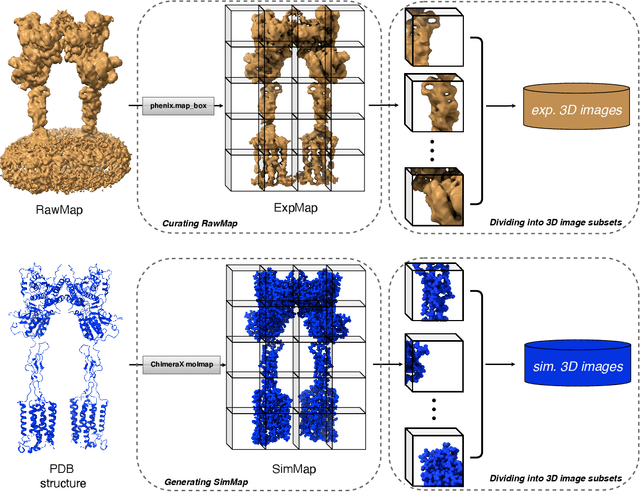
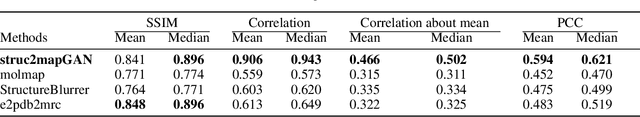
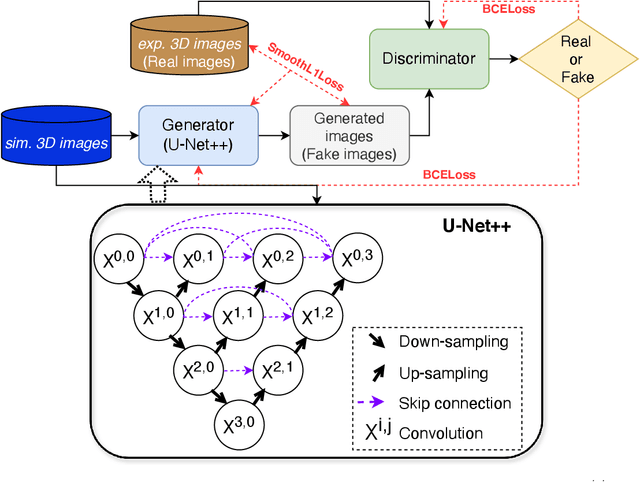
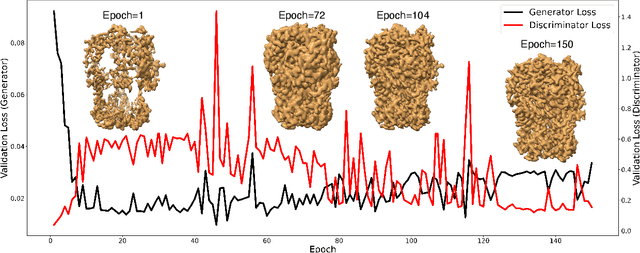
Generating synthetic cryogenic electron microscopy (cryo-EM) 3D density maps from molecular structures has potential important applications in structural biology. Yet existing simulation-based methods cannot mimic all the complex features present in experimental maps, such as secondary structure elements. As an alternative, we propose struc2mapGAN, a novel data-driven method that employs a generative adversarial network (GAN) to produce high-resolution experimental-like density maps from molecular structures. More specifically, struc2mapGAN uses a U-Net++ architecture as the generator, with an additional L1 loss term and further processing of raw experimental maps to enhance learning efficiency. While struc2mapGAN can promptly generate maps after training, we demonstrate that it outperforms existing simulation-based methods for a wide array of tested maps and across various evaluation metrics. Our code is available at https://github.com/chenwei-zhang/struc2mapGAN.
ViDa: Visualizing DNA hybridization trajectories with biophysics-informed deep graph embeddings
Nov 06, 2023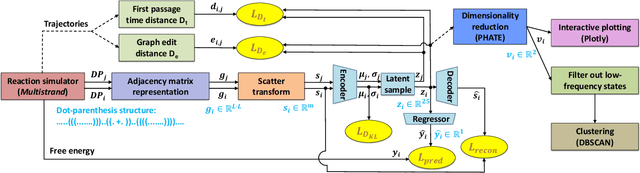

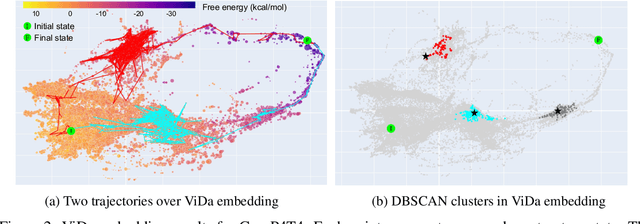

Visualization tools can help synthetic biologists and molecular programmers understand the complex reactive pathways of nucleic acid reactions, which can be designed for many potential applications and can be modelled using a continuous-time Markov chain (CTMC). Here we present ViDa, a new visualization approach for DNA reaction trajectories that uses a 2D embedding of the secondary structure state space underlying the CTMC model. To this end, we integrate a scattering transform of the secondary structure adjacency, a variational autoencoder, and a nonlinear dimensionality reduction method. We augment the training loss with domain-specific supervised terms that capture both thermodynamic and kinetic features. We assess ViDa on two well-studied DNA hybridization reactions. Our results demonstrate that the domain-specific features lead to significant quality improvements over the state-of-the-art in DNA state space visualization, successfully separating different folding pathways and thus providing useful insights into dominant reaction mechanisms.
Visualizing DNA reaction trajectories with deep graph embedding approaches
Nov 06, 2023


Synthetic biologists and molecular programmers design novel nucleic acid reactions, with many potential applications. Good visualization tools are needed to help domain experts make sense of the complex outputs of folding pathway simulations of such reactions. Here we present ViDa, a new approach for visualizing DNA reaction folding trajectories over the energy landscape of secondary structures. We integrate a deep graph embedding model with common dimensionality reduction approaches, to map high-dimensional data onto 2D Euclidean space. We assess ViDa on two well-studied and contrasting DNA hybridization reactions. Our preliminary results suggest that ViDa's visualization successfully separates trajectories with different folding mechanisms, thereby providing useful insight to users, and is a big improvement over the current state-of-the-art in DNA kinetics visualization.