 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConvolutional vs Large Language Models for Software Log Classification in Edge-Deployable Cellular Network Testing
Jul 04, 2024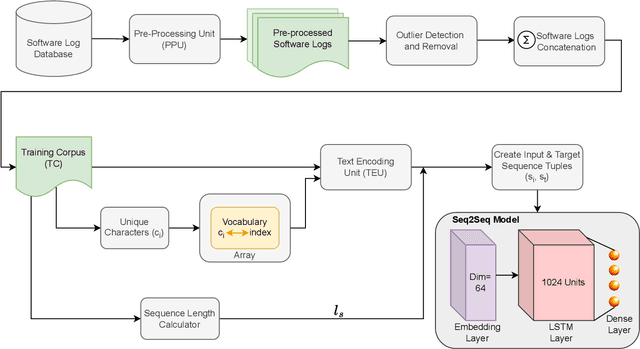
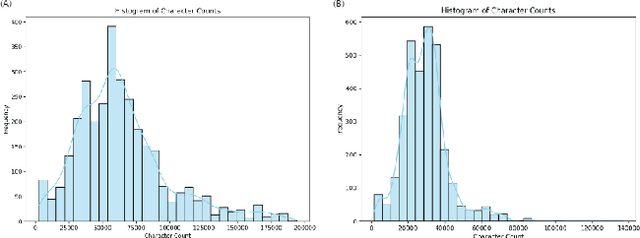
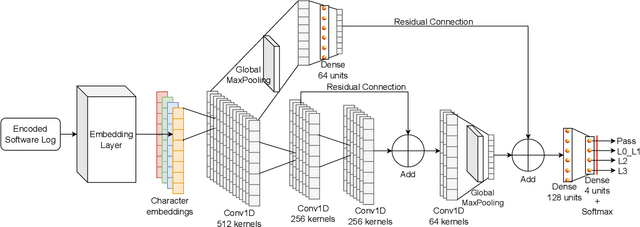
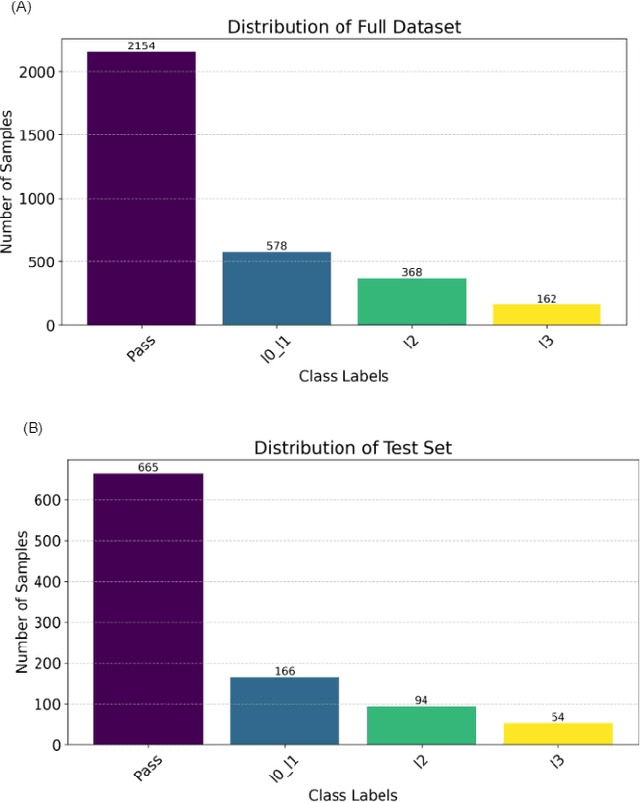
Software logs generated by sophisticated network emulators in the telecommunications industry, such as VIAVI TM500, are extremely complex, often comprising tens of thousands of text lines with minimal resemblance to natural language. Only specialised expert engineers can decipher such logs and troubleshoot defects in test runs. While AI offers a promising solution for automating defect triage, potentially leading to massive revenue savings for companies, state-of-the-art large language models (LLMs) suffer from significant drawbacks in this specialised domain. These include a constrained context window, limited applicability to text beyond natural language, and high inference costs. To address these limitations, we propose a compact convolutional neural network (CNN) architecture that offers a context window spanning up to 200,000 characters and achieves over 96% accuracy (F1>0.9) in classifying multifaceted software logs into various layers in the telecommunications protocol stack. Specifically, the proposed model is capable of identifying defects in test runs and triaging them to the relevant department, formerly a manual engineering process that required expert knowledge. We evaluate several LLMs; LLaMA2-7B, Mixtral 8x7B, Flan-T5, BERT and BigBird, and experimentally demonstrate their shortcomings in our specialized application. Despite being lightweight, our CNN significantly outperforms LLM-based approaches in telecommunications log classification while minimizing the cost of production. Our defect triaging AI model is deployable on edge devices without dedicated hardware and widely applicable across software logs in various industries.
Formula graph self-attention network for representation-domain independent materials discovery
Jan 14, 2022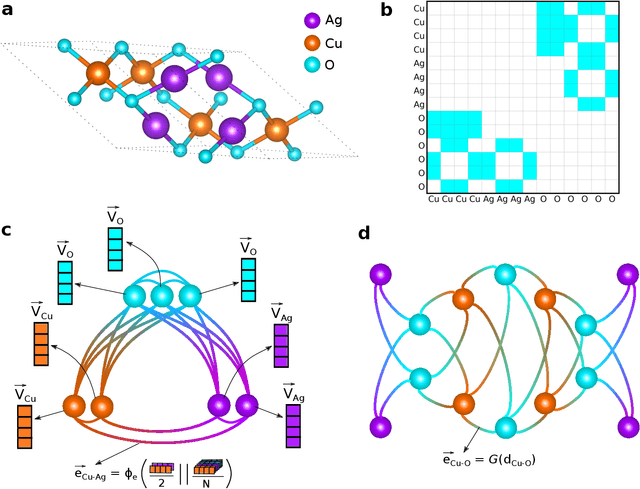
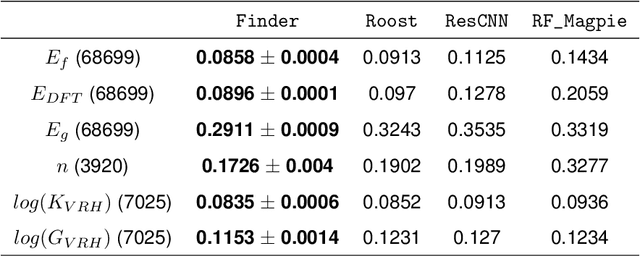
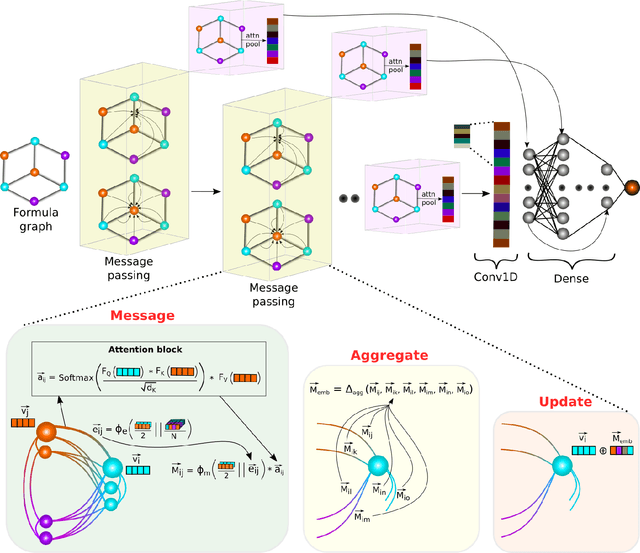
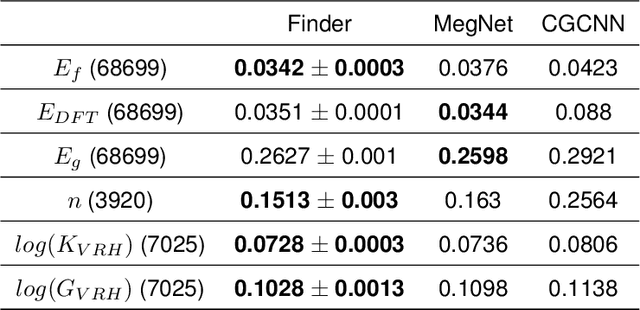
The success of machine learning (ML) in materials property prediction depends heavily on how the materials are represented for learning. Two dominant families of material descriptors exist, one that encodes crystal structure in the representation and the other that only uses stoichiometric information with the hope of discovering new materials. Graph neural networks (GNNs) in particular have excelled in predicting material properties within chemical accuracy. However, current GNNs are limited to only one of the above two avenues owing to the little overlap between respective material representations. Here, we introduce a new concept of formula graph which unifies both stoichiometry-only and structure-based material descriptors. We further develop a self-attention integrated GNN that assimilates a formula graph and show that the proposed architecture produces material embeddings transferable between the two domains. Our model substantially outperforms previous structure-based GNNs as well as structure-agnostic counterparts while exhibiting better sample efficiency and faster convergence. Finally, the model is applied in a challenging exemplar to predict the complex dielectric function of materials and nominate new substances that potentially exhibit epsilon-near-zero phenomena.
Analogical discovery of disordered perovskite oxides by crystal structure information hidden in unsupervised material fingerprints
May 25, 2021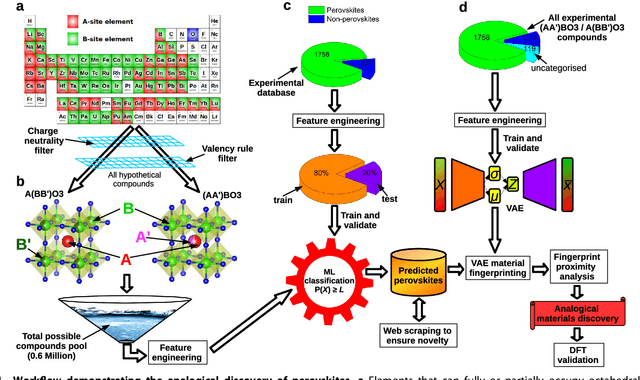
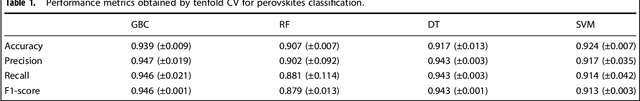
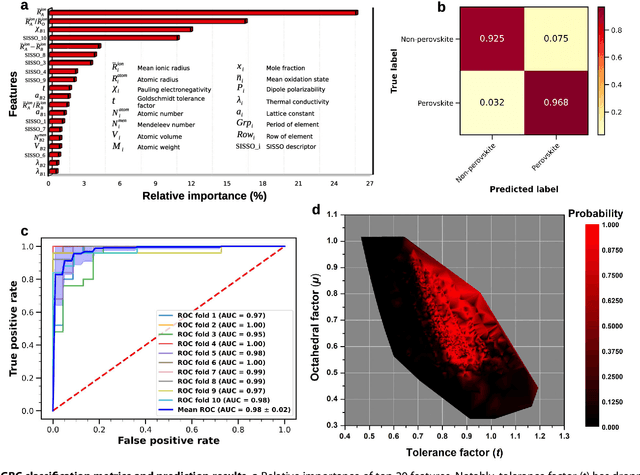
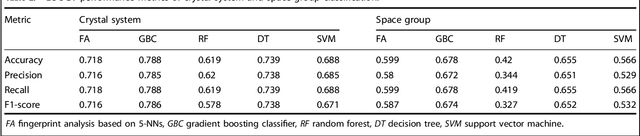
Compositional disorder induces myriad captivating phenomena in perovskites. Target-driven discovery of perovskite solid solutions has been a great challenge due to the analytical complexity introduced by disorder. Here, we demonstrate that an unsupervised deep learning strategy can find fingerprints of disordered materials that embed perovskite formability and underlying crystal structure information by learning only from the chemical composition, manifested in (A1-xA'x)BO3 and A(B1-xB'x)O3 formulae. This phenomenon can be capitalized to predict the crystal symmetry of experimental compositions, outperforming several supervised machine learning (ML) algorithms. The educated nature of material fingerprints has led to the conception of analogical materials discovery that facilitates inverse exploration of promising perovskites based on similarity investigation with known materials. The search space of unstudied perovskites is screened from ~600,000 feasible compounds using experimental data powered ML models and automated web mining tools at a 94% success rate. This concept further provides insights on possible phase transitions and computational modelling of complex compositions. The proposed quantitative analysis of materials analogies is expected to bridge the gap between the existing materials literature and the undiscovered terrain.