 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFormula graph self-attention network for representation-domain independent materials discovery
Paper and Code
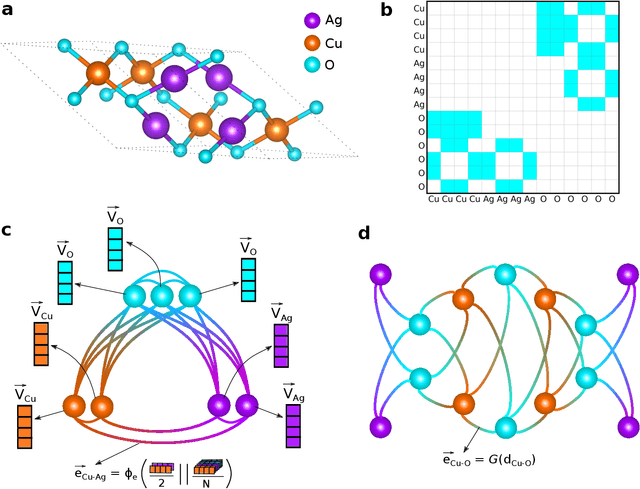
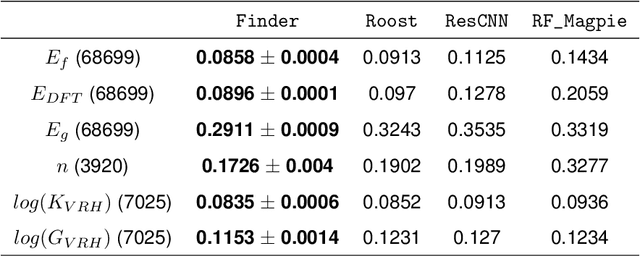
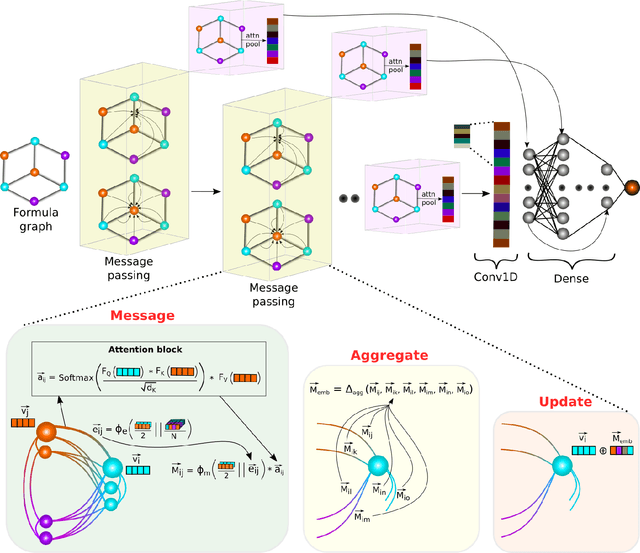
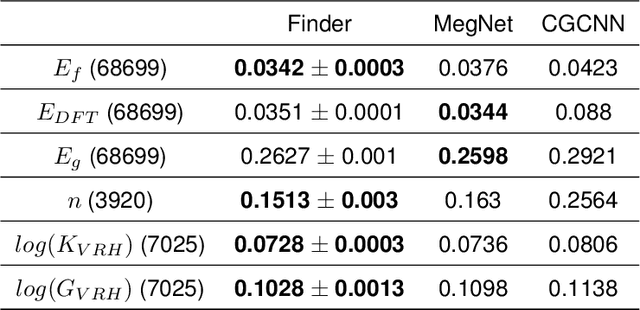
The success of machine learning (ML) in materials property prediction depends heavily on how the materials are represented for learning. Two dominant families of material descriptors exist, one that encodes crystal structure in the representation and the other that only uses stoichiometric information with the hope of discovering new materials. Graph neural networks (GNNs) in particular have excelled in predicting material properties within chemical accuracy. However, current GNNs are limited to only one of the above two avenues owing to the little overlap between respective material representations. Here, we introduce a new concept of formula graph which unifies both stoichiometry-only and structure-based material descriptors. We further develop a self-attention integrated GNN that assimilates a formula graph and show that the proposed architecture produces material embeddings transferable between the two domains. Our model substantially outperforms previous structure-based GNNs as well as structure-agnostic counterparts while exhibiting better sample efficiency and faster convergence. Finally, the model is applied in a challenging exemplar to predict the complex dielectric function of materials and nominate new substances that potentially exhibit epsilon-near-zero phenomena.