 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhich Hyperparameters to Optimise? An Investigation of Evolutionary Hyperparameter Optimisation in Graph Neural Network For Molecular Property Prediction
Paper and Code
Apr 14, 2021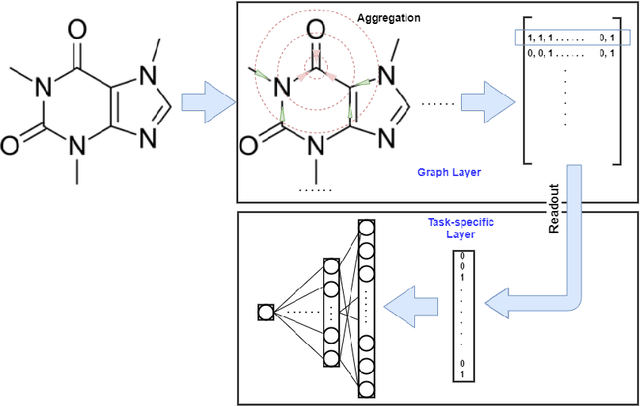

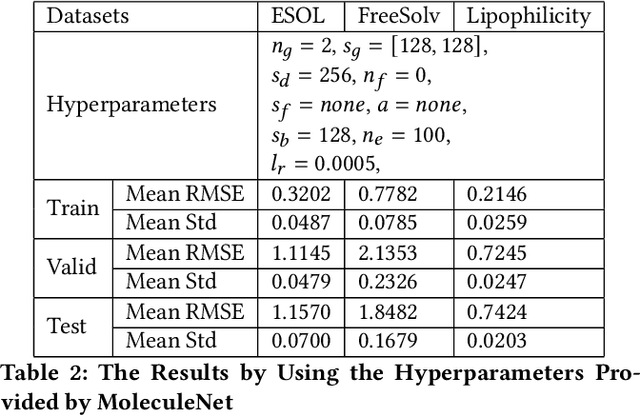
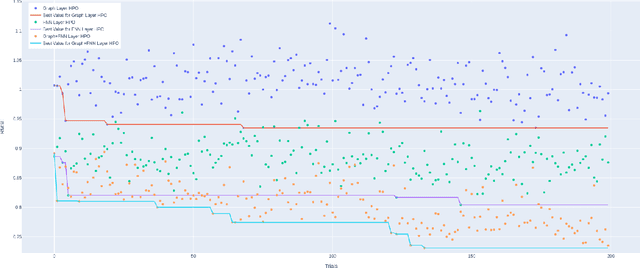
Recently, the study of graph neural network (GNN) has attracted much attention and achieved promising performance in molecular property prediction. Most GNNs for molecular property prediction are proposed based on the idea of learning the representations for the nodes by aggregating the information of their neighbor nodes (e.g. atoms). Then, the representations can be passed to subsequent layers to deal with individual downstream tasks. Therefore, the architectures of GNNs can be considered as being composed of two core parts: graph-related layers and task-specific layers. Facing real-world molecular problems, the hyperparameter optimization for those layers are vital. Hyperparameter optimization (HPO) becomes expensive in this situation because evaluating candidate solutions requires massive computational resources to train and validate models. Furthermore, a larger search space often makes the HPO problems more challenging. In this research, we focus on the impact of selecting two types of GNN hyperparameters, those belonging to graph-related layers and those of task-specific layers, on the performance of GNN for molecular property prediction. In our experiments. we employed a state-of-the-art evolutionary algorithm (i.e., CMA-ES) for HPO. The results reveal that optimizing the two types of hyperparameters separately can gain the improvements on GNNs' performance, but optimising both types of hyperparameters simultaneously will lead to predominant improvements. Meanwhile, our study also further confirms the importance of HPO for GNNs in molecular property prediction problems.