 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTight basis cycle representatives for persistent homology of large data sets
Paper and Code
Jun 06, 2022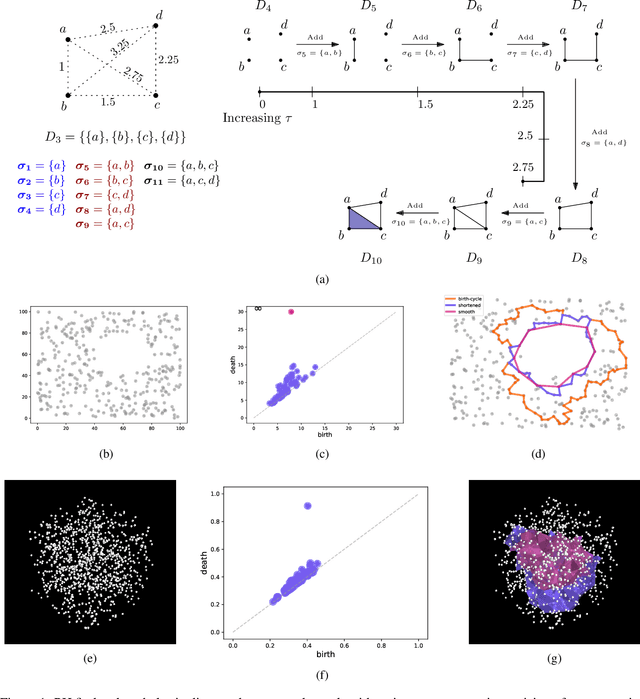

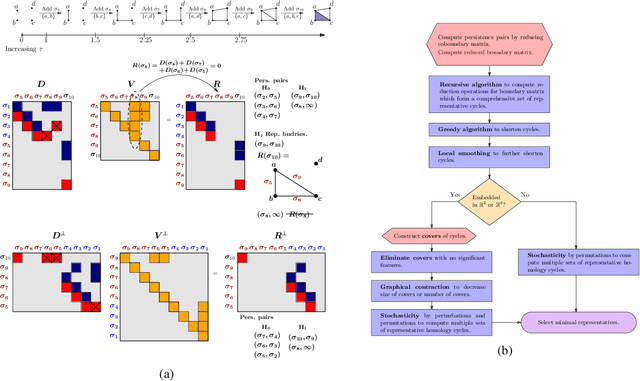
Persistent homology (PH) is a popular tool for topological data analysis that has found applications across diverse areas of research. It provides a rigorous method to compute robust topological features in discrete experimental observations that often contain various sources of uncertainties. Although powerful in theory, PH suffers from high computation cost that precludes its application to large data sets. Additionally, most analyses using PH are limited to computing the existence of nontrivial features. Precise localization of these features is not generally attempted because, by definition, localized representations are not unique and because of even higher computation cost. For scientific applications, such a precise location is a sine qua non for determining functional significance. Here, we provide a strategy and algorithms to compute tight representative boundaries around nontrivial robust features in large data sets. To showcase the efficiency of our algorithms and the precision of computed boundaries, we analyze three data sets from different scientific fields. In the human genome, we found an unexpected effect on loops through chromosome 13 and the sex chromosomes, upon impairment of chromatin loop formation. In a distribution of galaxies in the universe, we found statistically significant voids. In protein homologs with significantly different topology, we found voids attributable to ligand-interaction, mutation, and differences between species.