 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTaking a Respite from Representation Learning for Molecular Property Prediction
Paper and Code
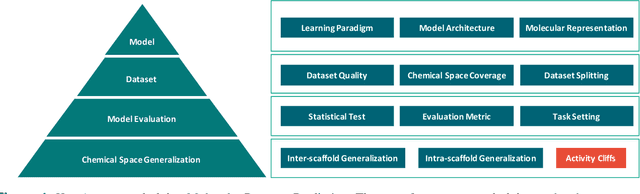
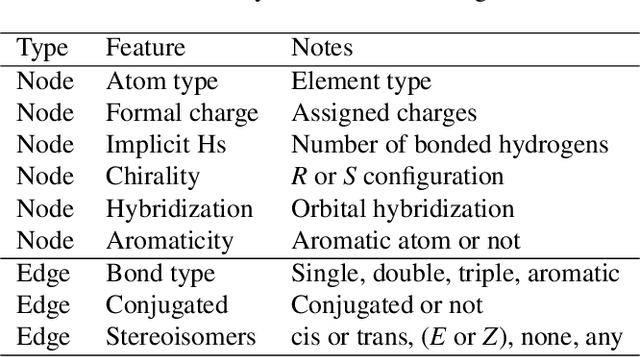
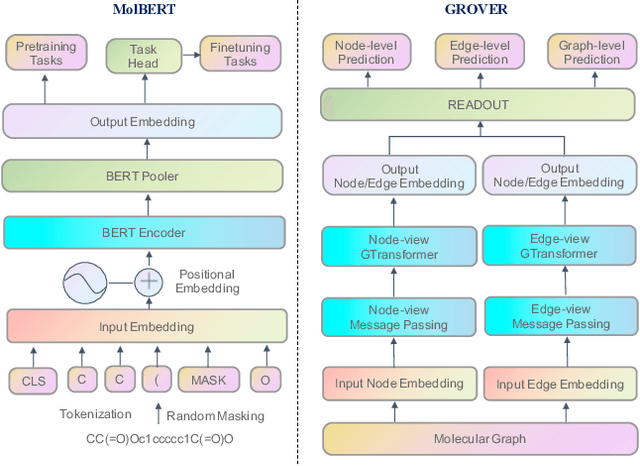

Artificial intelligence (AI) has been widely applied in drug discovery with a major task as molecular property prediction. Despite the boom of AI techniques in molecular representation learning, some key aspects underlying molecular property prediction haven't been carefully examined yet. In this study, we conducted a systematic comparison on three representative models, random forest, MolBERT and GROVER, which utilize three major molecular representations, extended-connectivity fingerprints, SMILES strings and molecular graphs, respectively. Notably, MolBERT and GROVER, are pretrained on large-scale unlabelled molecule corpuses in a self-supervised manner. In addition to the commonly used MoleculeNet benchmark datasets, we also assembled a suite of opioids-related datasets for downstream prediction evaluation. We first conducted dataset profiling on label distribution and structural analyses; we also examined the activity cliffs issue in the opioids-related datasets. Then, we trained 4,320 predictive models and evaluated the usefulness of the learned representations. Furthermore, we explored into the model evaluation by studying the effect of statistical tests, evaluation metrics and task settings. Finally, we dissected the chemical space generalization into inter-scaffold and intra-scaffold generalization and measured prediction performance to evaluate model generalizbility under both settings. By taking this respite, we reflected on the key aspects underlying molecular property prediction, the awareness of which can, hopefully, bring better AI techniques in this field.