 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of the electron density of states for crystalline compounds with Atomistic Line Graph Neural Networks
Paper and Code
Jan 20, 2022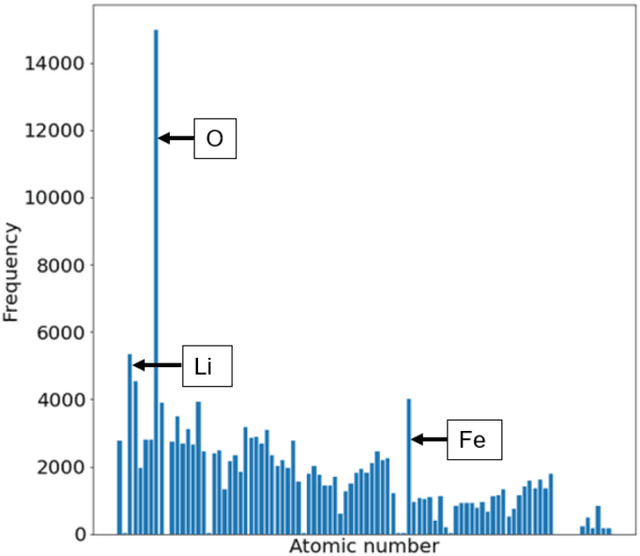
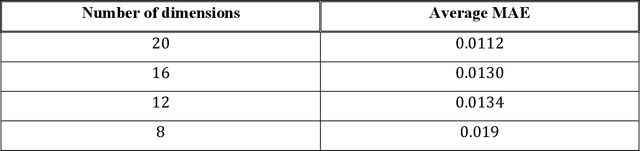
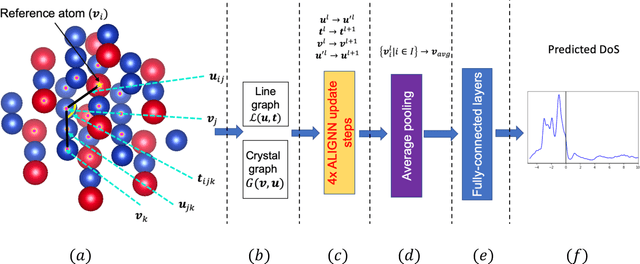
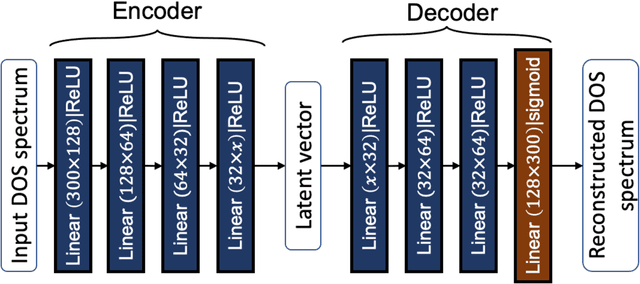
Machine learning (ML) based models have greatly enhanced the traditional materials discovery and design pipeline. Specifically, in recent years, surrogate ML models for material property prediction have demonstrated success in predicting discrete scalar-valued target properties to within reasonable accuracy of their DFT-computed values. However, accurate prediction of spectral targets such as the electron Density of States (DOS) poses a much more challenging problem due to the complexity of the target, and the limited amount of available training data. In this study, we present an extension of the recently developed Atomistic Line Graph Neural Network (ALIGNN) to accurately predict DOS of a large set of material unit cell structures, trained to the publicly available JARVIS-DFT dataset. Furthermore, we evaluate two methods of representation of the target quantity - a direct discretized spectrum, and a compressed low-dimensional representation obtained using an autoencoder. Through this work, we demonstrate the utility of graph-based featurization and modeling methods in the prediction of complex targets that depend on both chemistry and directional characteristics of material structures.