 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeasuring the Similarity between Materials with an Emphasis on the Materials Distinctiveness
Paper and Code
Mar 23, 2019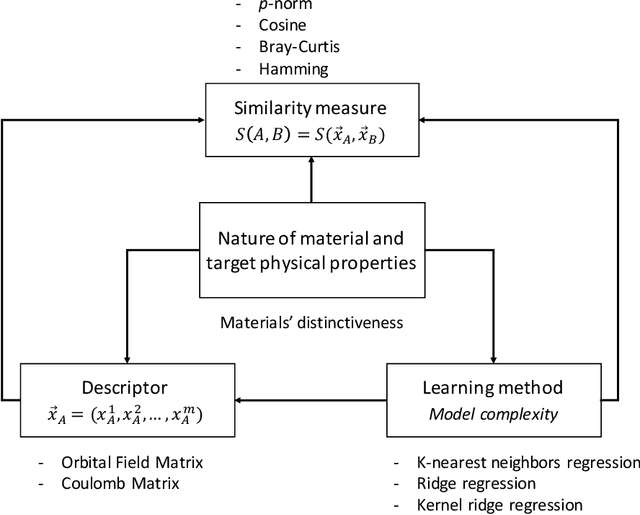
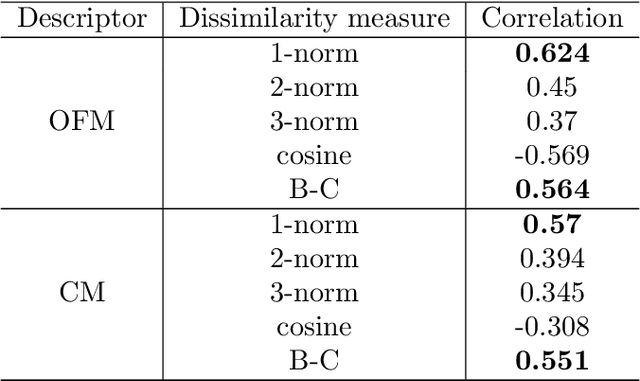
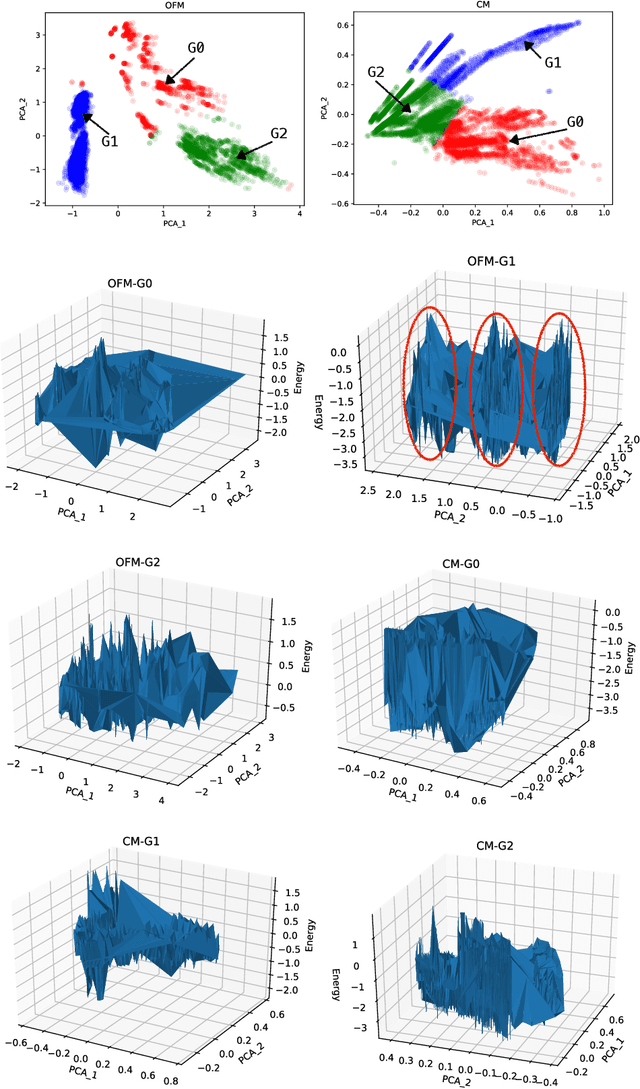

In this study, we establish a basis for selecting similarity measures when applying machine learning techniques to solve materials science problems. This selection is considered with an emphasis on the distinctiveness between materials that reflect their nature well. We perform a case study with a dataset of rare-earth transition metal crystalline compounds represented using the Orbital Field Matrix descriptor and the Coulomb Matrix descriptor. We perform predictions of the formation energies using k-nearest neighbors regression, ridge regression, and kernel ridge regression. Through detailed analyses of the yield prediction accuracy, we examine the relationship between the characteristics of the material representation and similarity measures, and the complexity of the energy function they can capture. Empirical experiments and theoretical analysis reveal that similarity measures and kernels that minimize the loss of materials distinctiveness improve the prediction performance.