 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKnowledge Discovery from Atomic Structures using Feature Importances
Paper and Code
Feb 28, 2023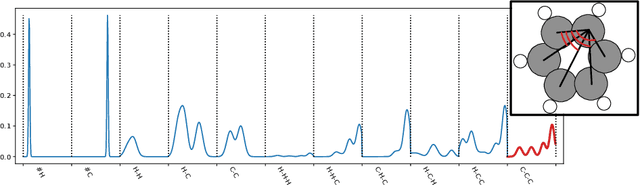
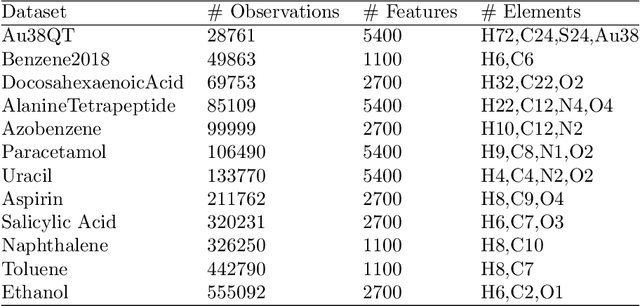
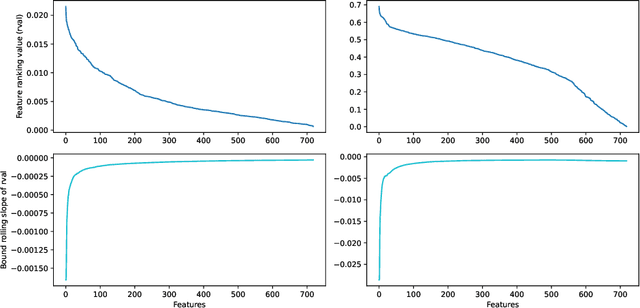
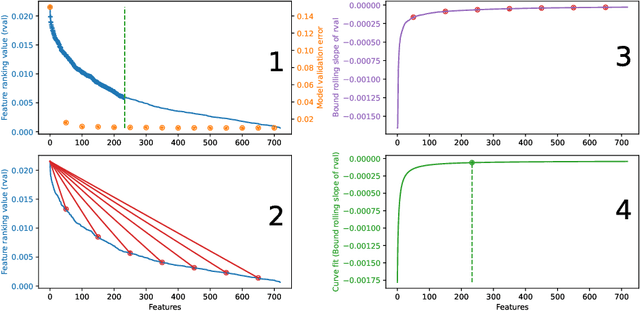
Molecular-level understanding of the interactions between the constituents of an atomic structure is essential for designing novel materials in various applications. This need goes beyond the basic knowledge of the number and types of atoms, their chemical composition, and the character of the chemical interactions. The bigger picture takes place on the quantum level which can be addressed by using the Density-functional theory (DFT). Use of DFT, however, is a computationally taxing process, and its results do not readily provide easily interpretable insight into the atomic interactions which would be useful information in material design. An alternative way to address atomic interactions is to use an interpretable machine learning approach, where a predictive DFT surrogate is constructed and analyzed. The purpose of this paper is to propose such a procedure using a modification of the recently published interpretable distance-based regression method. Our tests with a representative benchmark set of molecules and a complex hybrid nanoparticle confirm the viability and usefulness of the proposed approach.