Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigh Performance of Gradient Boosting in Binding Affinity Prediction
Paper and Code
May 14, 2022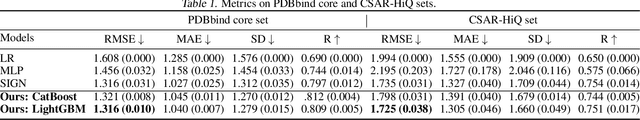
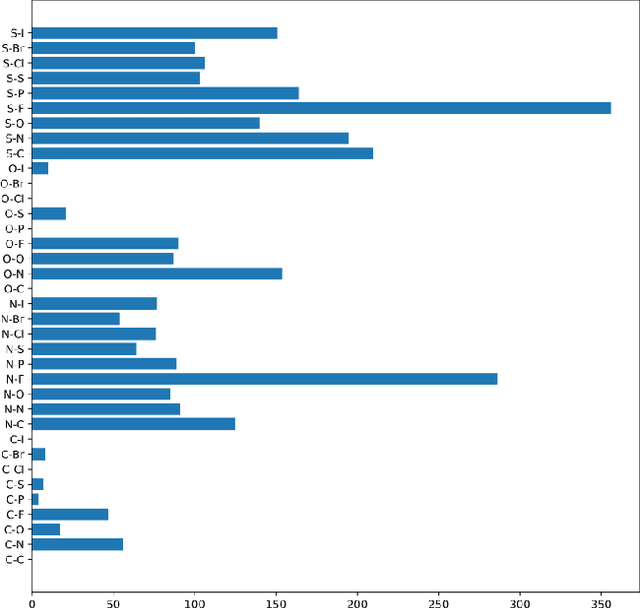
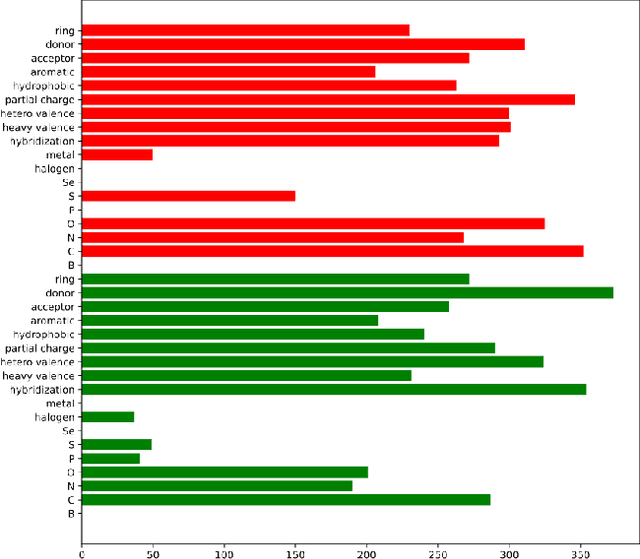
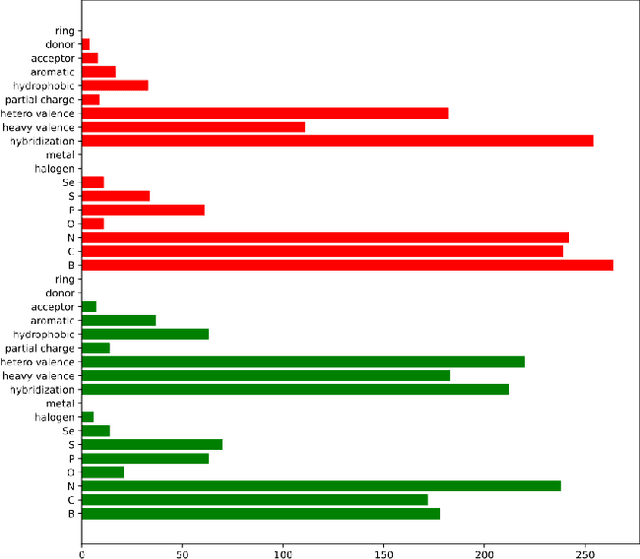
Prediction of protein-ligand (PL) binding affinity remains the key to drug discovery. Popular approaches in recent years involve graph neural networks (GNNs), which are used to learn the topology and geometry of PL complexes. However, GNNs are computationally heavy and have poor scalability to graph sizes. On the other hand, traditional machine learning (ML) approaches, such as gradient-boosted decision trees (GBDTs), are lightweight yet extremely efficient for tabular data. We propose to use PL interaction features along with PL graph-level features in GBDT. We show that this combination outperforms the existing solutions.
View paper on