 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Network Architecture Search for Molecular Property Prediction
Paper and Code
Aug 27, 2020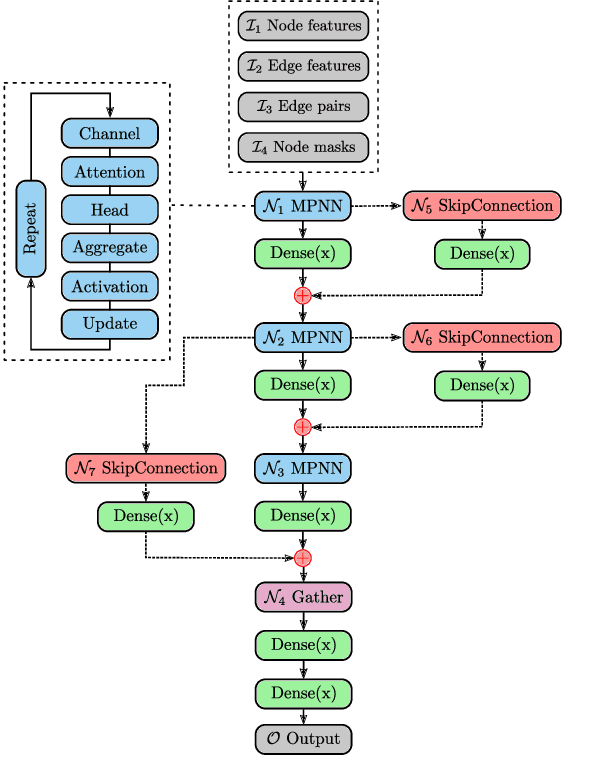

Predicting the properties of a molecule from its structure is a challenging task. Recently, deep learning methods have improved the state of the art for this task because of their ability to learn useful features from the given data. By treating molecule structure as graphs, where atoms and bonds are modeled as nodes and edges, graph neural networks (GNNs) have been widely used to predict molecular properties. However, the design and development of GNNs for a given data set rely on labor-intensive design and tuning of the network architectures. Neural architecture search (NAS) is a promising approach to discover high-performing neural network architectures automatically. To that end, we develop an NAS approach to automate the design and development of GNNs for molecular property prediction. Specifically, we focus on automated development of message-passing neural networks (MPNNs) to predict the molecular properties of small molecules in quantum mechanics and physical chemistry data sets from the MoleculeNet benchmark. We demonstrate the superiority of the automatically discovered MPNNs by comparing them with manually designed GNNs from the MoleculeNet benchmark. We study the relative importance of the choices in the MPNN search space, demonstrating that customizing the architecture is critical to enhancing performance in molecular property prediction and that the proposed approach can perform customization automatically with minimal manual effort.