 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGemNet: Universal Directional Graph Neural Networks for Molecules
Paper and Code
Jun 22, 2021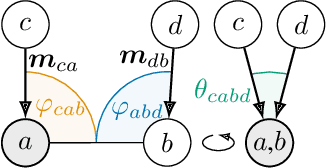


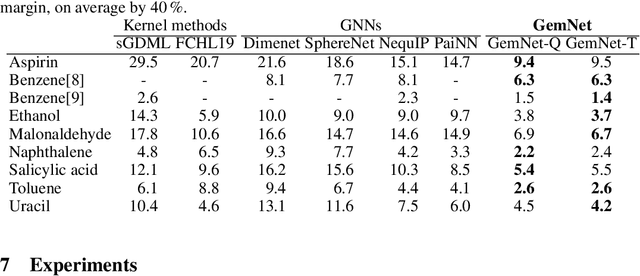
Effectively predicting molecular interactions has the potential to accelerate molecular dynamics by multiple orders of magnitude and thus revolutionize chemical simulations. Graph neural networks (GNNs) have recently shown great successes for this task, overtaking classical methods based on fixed molecular kernels. However, they still appear very limited from a theoretical perspective, since regular GNNs cannot distinguish certain types of graphs. In this work we close this gap between theory and practice. We show that GNNs with directed edge embeddings and two-hop message passing are indeed universal approximators for predictions that are invariant to global rotation and translation, and equivariant to permutation. We then leverage these insights and multiple structural improvements to propose the geometric message passing neural network (GemNet). We demonstrate the benefits of the proposed changes in multiple ablation studies. GemNet outperforms previous models on the COLL and MD17 molecular dynamics datasets by 34% and 40%, performing especially well on the most challenging molecules.