 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFragment-based Pretraining and Finetuning on Molecular Graphs
Paper and Code
Oct 05, 2023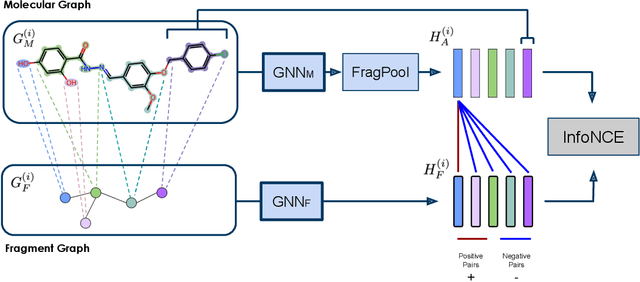
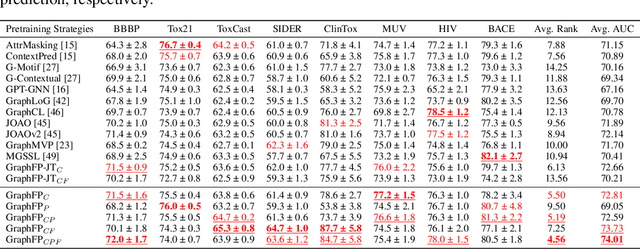
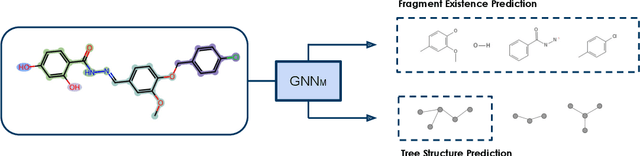
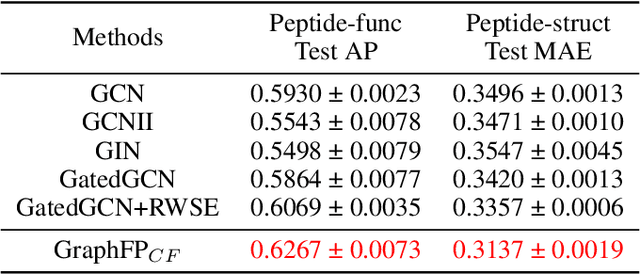
Property prediction on molecular graphs is an important application of Graph Neural Networks (GNNs). Recently, unlabeled molecular data has become abundant, which facilitates the rapid development of self-supervised learning for GNNs in the chemical domain. In this work, we propose pretraining GNNs at the fragment level, which serves as a promising middle ground to overcome the limitations of node-level and graph-level pretraining. Borrowing techniques from recent work on principle subgraph mining, we obtain a compact vocabulary of prevalent fragments that span a large pretraining dataset. From the extracted vocabulary, we introduce several fragment-based contrastive and predictive pretraining tasks. The contrastive learning task jointly pretrains two different GNNs: one based on molecular graphs and one based on fragment graphs, which represents high-order connectivity within molecules. By enforcing the consistency between the fragment embedding and the aggregated embedding of the corresponding atoms from the molecular graphs, we ensure that both embeddings capture structural information at multiple resolutions. The structural information of the fragment graphs is further exploited to extract auxiliary labels for the graph-level predictive pretraining. We employ both the pretrained molecular-based and fragment-based GNNs for downstream prediction, thus utilizing the fragment information during finetuning. Our models advance the performances on 5 out of 8 common molecular benchmarks and improve the performances on long-range biological benchmarks by at least 11.5%.