 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFoldingZero: Protein Folding from Scratch in Hydrophobic-Polar Model
Paper and Code
Dec 03, 2018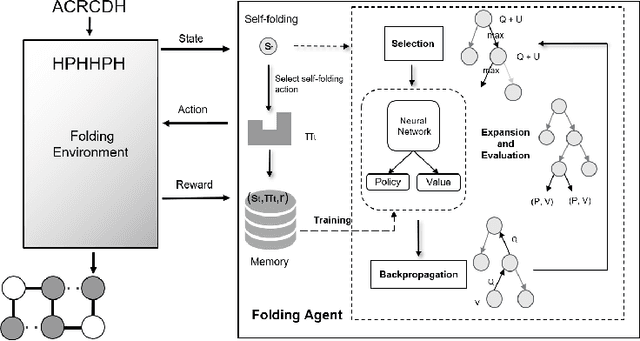
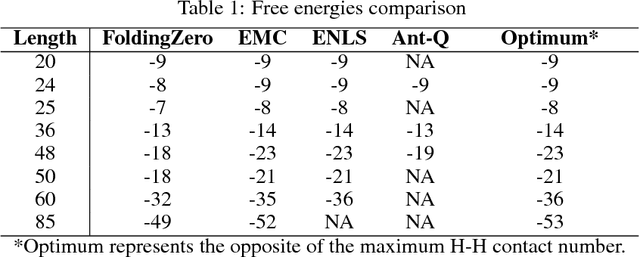
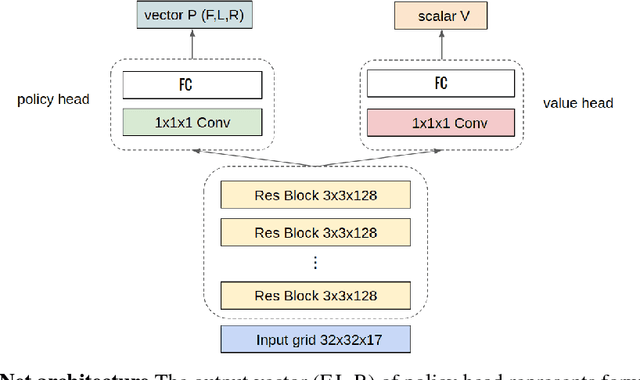
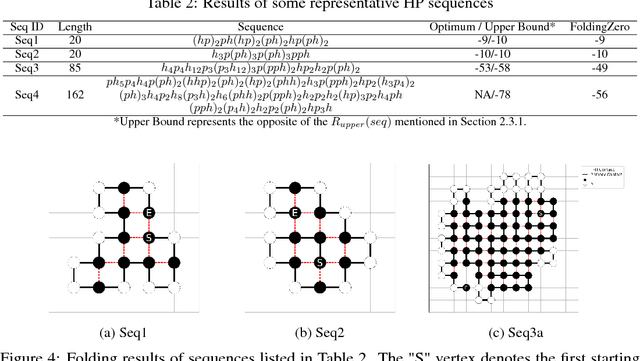
De novo protein structure prediction from amino acid sequence is one of the most challenging problems in computational biology. As one of the extensively explored mathematical models for protein folding, Hydrophobic-Polar (HP) model enables thorough investigation of protein structure formation and evolution. Although HP model discretizes the conformational space and simplifies the folding energy function, it has been proven to be an NP-complete problem. In this paper, we propose a novel protein folding framework FoldingZero, self-folding a de novo protein 2D HP structure from scratch based on deep reinforcement learning. FoldingZero features the coupled approach of a two-head (policy and value heads) deep convolutional neural network (HPNet) and a regularized Upper Confidence Bounds for Trees (R-UCT). It is trained solely by a reinforcement learning algorithm, which improves HPNet and R-UCT iteratively through iterative policy optimization. Without any supervision and domain knowledge, FoldingZero not only achieves comparable results, but also learns the latent folding knowledge to stabilize the structure. Without exponential computation, FoldingZero shows promising potential to be adopted for real-world protein properties prediction.