 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeClassifying COVID-19 Spike Sequences from Geographic Location Using Deep Learning
Paper and Code
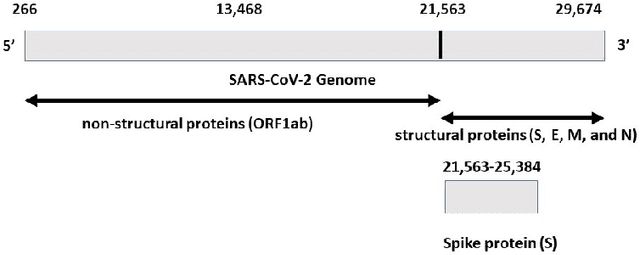
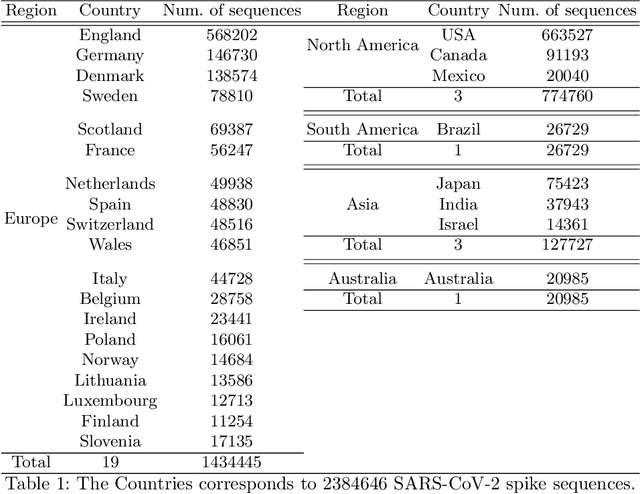
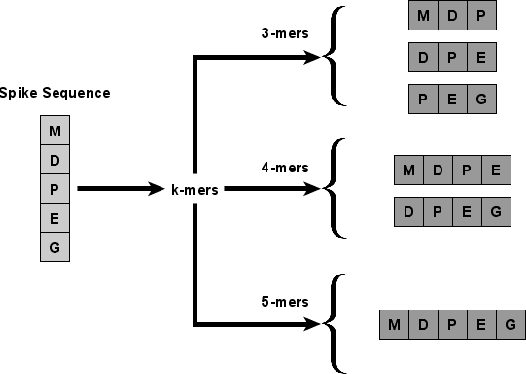
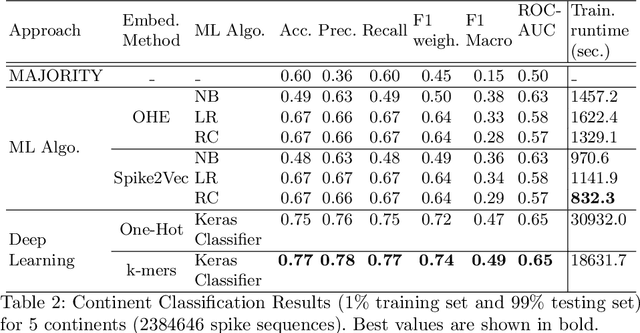
With the rapid spread of COVID-19 worldwide, viral genomic data is available in the order of millions of sequences on public databases such as GISAID. This \emph{Big Data} creates a unique opportunity for analysis towards the research of effective vaccine development for current pandemics, and avoiding or mitigating future pandemics. One piece of information that comes with every such viral sequence is the geographical location where it was collected -- the patterns found between viral variants and geographic location surely being an important part of this analysis. One major challenge that researchers face is processing such huge, highly dimensional data to get useful insights as quickly as possible. Most of the existing methods face scalability issues when dealing with the magnitude of such data. In this paper, we propose an algorithm that first computes a numerical representation of the spike protein sequence of SARS-CoV-2 using $k$-mers substrings) and then uses a deep learning-based model to classify the sequences in terms of geographical location. We show that our proposed model significantly outperforms the baselines. We also show the importance of different amino acids in the spike sequences by computing the information gain corresponding to the true class labels.