 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCatalyst design using actively learned machine with non-ab initio input features towards CO2 reduction reactions
Paper and Code
Sep 14, 2017
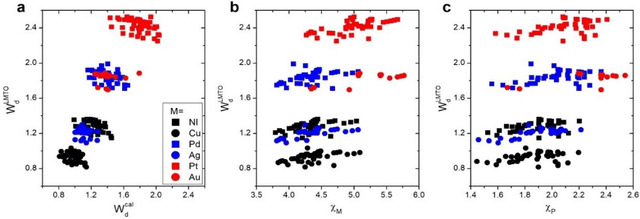
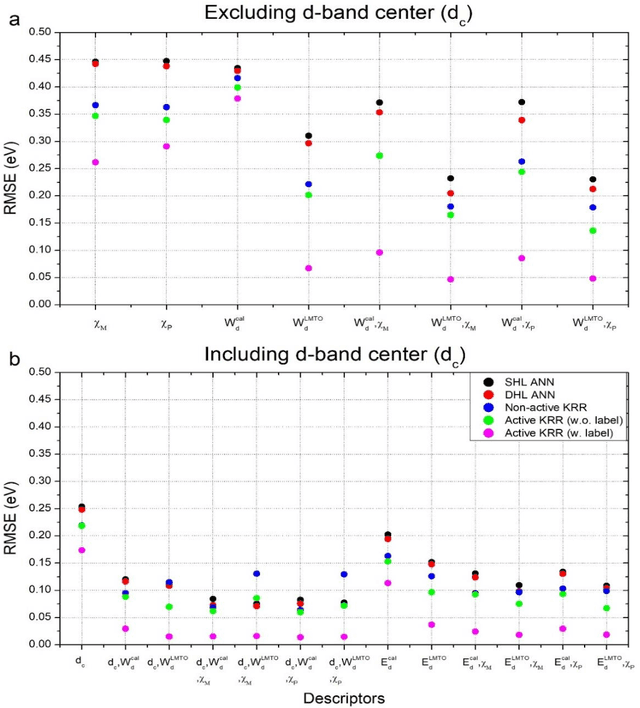
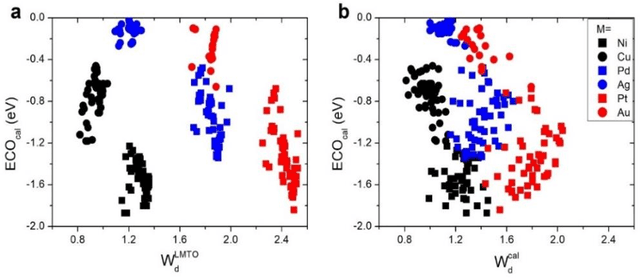
In conventional chemisorption model, the d-band center theory (augmented sometimes with the upper edge of d-band for imporved accuarcy) plays a central role in predicting adsorption energies and catalytic activity as a function of d-band center of the solid surfaces, but it requires density functional calculations that can be quite costly for large scale screening purposes of materials. In this work, we propose to use the d-band width of the muffin-tin orbital theory (to account for local coordination environment) plus electronegativity (to account for adsorbate renormalization) as a simple set of alternative descriptors for chemisorption, which do not demand the ab initio calculations. This pair of descriptors are then combined with machine learning methods, namely, artificial neural network (ANN) and kernel ridge regression (KRR), to allow large scale materials screenings. We show, for a toy set of 263 alloy systems, that the CO adsorption energy can be predicted with a remarkably small mean absolute deviation error of 0.05 eV, a significantly improved result as compared to 0.13 eV obtained with descriptors including costly d-band center calculations in literature. We achieved this high accuracy by utilizing an active learning algorithm, without which the accuracy was 0.18 eV otherwise. As a practical application of this machine, we identified Cu3Y@Cu as a highly active and cost-effective electrochemical CO2 reduction catalyst to produce CO with the overpotential 0.37 V lower than Au catalyst.