 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBoundary-Guided Learning for Gene Expression Prediction in Spatial Transcriptomics
Paper and Code
Dec 05, 2024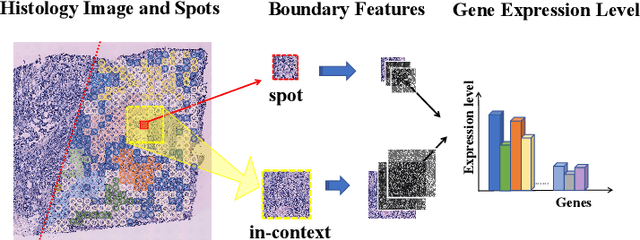
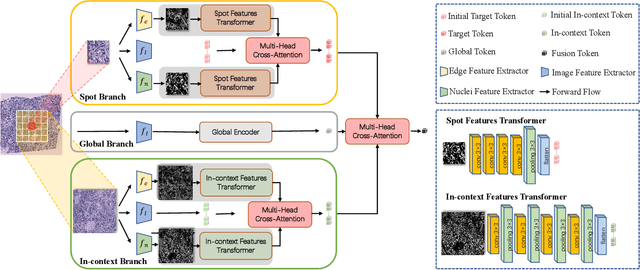
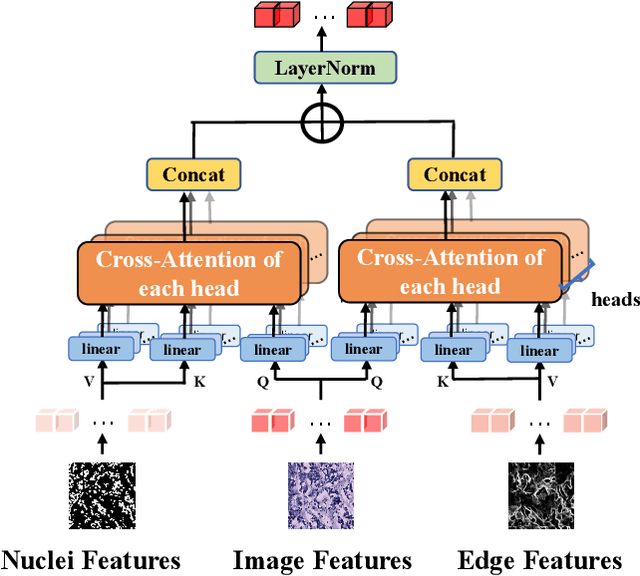
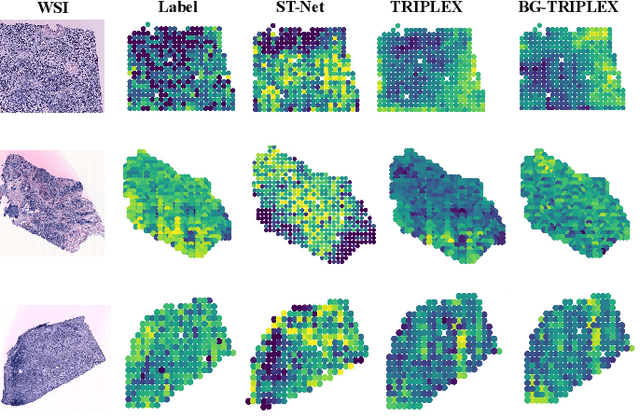
Spatial transcriptomics (ST) has emerged as an advanced technology that provides spatial context to gene expression. Recently, deep learning-based methods have shown the capability to predict gene expression from WSI data using ST data. Existing approaches typically extract features from images and the neighboring regions using pretrained models, and then develop methods to fuse this information to generate the final output. However, these methods often fail to account for the cellular structure similarity, cellular density and the interactions within the microenvironment. In this paper, we propose a framework named BG-TRIPLEX, which leverages boundary information extracted from pathological images as guiding features to enhance gene expression prediction from WSIs. Specifically, our model consists of three branches: the spot, in-context and global branches. In the spot and in-context branches, boundary information, including edge and nuclei characteristics, is extracted using pretrained models. These boundary features guide the learning of cellular morphology and the characteristics of microenvironment through Multi-Head Cross-Attention. Finally, these features are integrated with global features to predict the final output. Extensive experiments were conducted on three public ST datasets. The results demonstrate that our BG-TRIPLEX consistently outperforms existing methods in terms of Pearson Correlation Coefficient (PCC). This method highlights the crucial role of boundary features in understanding the complex interactions between WSI and gene expression, offering a promising direction for future research.