 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeALPHAGMUT: A Rationale-Guided Alpha Shape Graph Neural Network to Evaluate Mutation Effects
Paper and Code
Jun 13, 2024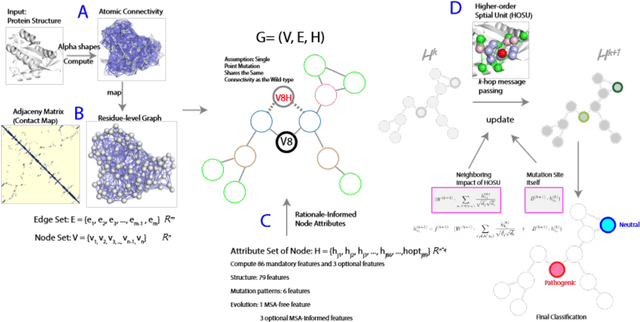



In silico methods evaluating the mutation effects of missense mutations are providing an important approach for understanding mutations in personal genomes and identifying disease-relevant biomarkers. However, existing methods, including deep learning methods, heavily rely on sequence-aware information, and do not fully leverage the potential of available 3D structural information. In addition, these methods may exhibit an inability to predict mutations in domains difficult to formulate sequence-based embeddings. In this study, we introduce a novel rationale-guided graph neural network AlphaGMut to evaluate mutation effects and to distinguish pathogenic mutations from neutral mutations. We compute the alpha shapes of protein structures to obtain atomic-resolution edge connectivities and map them to an accurate residue-level graph representation. We then compute structural-, topological-, biophysical-, and sequence properties of the mutation sites, which are assigned as node attributes in the graph. These node attributes could effectively guide the graph neural network to learn the difference between pathogenic and neutral mutations using k-hop message passing with a short training period. We demonstrate that AlphaGMut outperforms state-of-the-art methods, including DeepMind's AlphaMissense, in many performance metrics. In addition, AlphaGMut has the advantage of performing well in alignment-free settings, which provides broader prediction coverage and better generalization compared to current methods requiring deep sequence-aware information.