 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Systematic Comparison Study on Hyperparameter Optimisation of Graph Neural Networks for Molecular Property Prediction
Paper and Code
Feb 08, 2021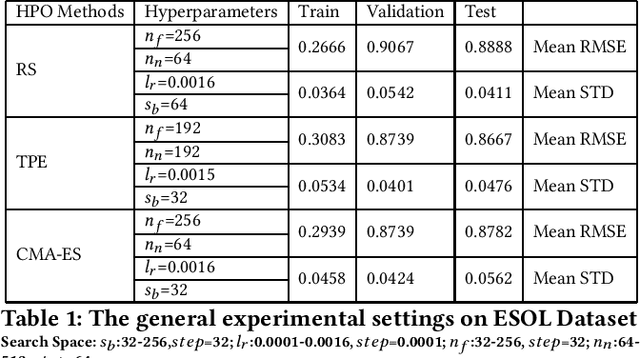
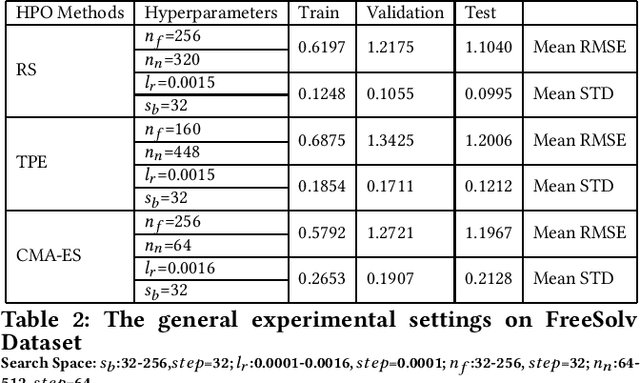
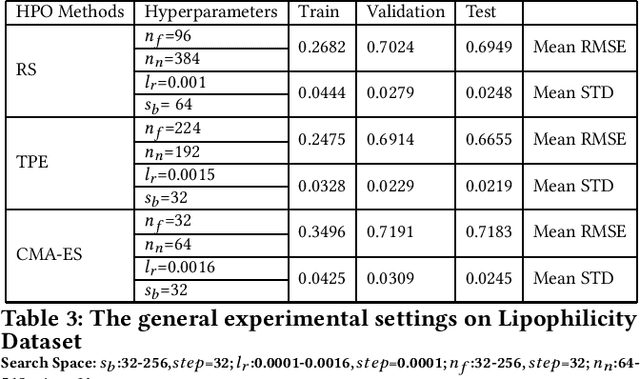
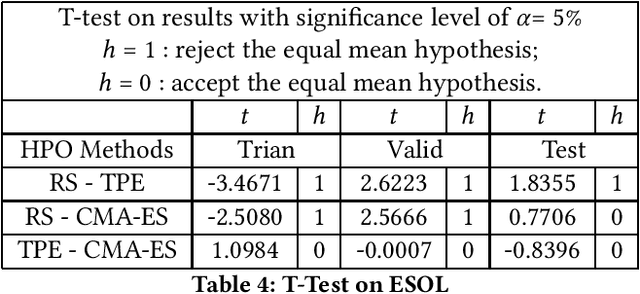
Graph neural networks (GNNs) have been proposed for a wide range of graph-related learning tasks. In particular, in recent years there has been an increasing number of GNN systems that were applied to predict molecular properties. However, in theory, there are infinite choices of hyperparameter settings for GNNs, and a direct impediment is to select appropriate hyperparameters to achieve satisfactory performance with lower computational cost. Meanwhile, the sizes of many molecular datasets are far smaller than many other datasets in typical deep learning applications, and most hyperparameter optimization (HPO) methods have not been explored in terms of their efficiencies on such small datasets in molecular domain. In this paper, we conducted a theoretical analysis of common and specific features for two state-of-the-art and popular algorithms for HPO: TPE and CMA-ES, and we compared them with random search (RS), which is used as a baseline. Experimental studies are carried out on several benchmarks in MoleculeNet, from different perspectives to investigate the impact of RS, TPE, and CMA-ES on HPO of GNNs for molecular property prediction. In our experiments, we concluded that RS, TPE, and CMA-ES have their individual advantages in tackling different specific molecular problems. Finally, we believe our work will motivate further research on GNN as applied to molecular machine learning problems in chemistry and materials sciences.