 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA study of structural properties on profiles HMMs
Paper and Code
Dec 11, 2008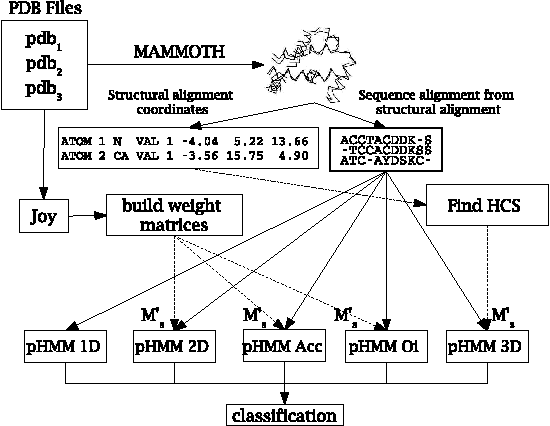
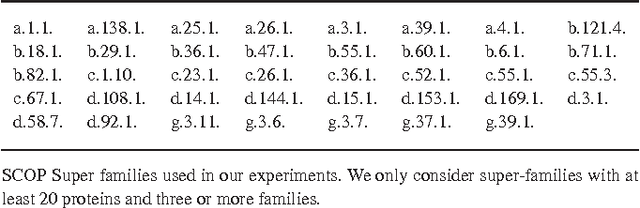
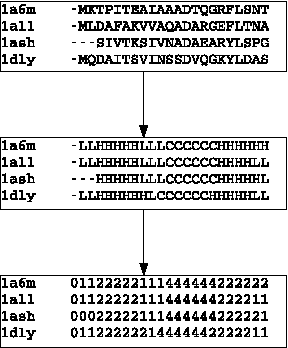
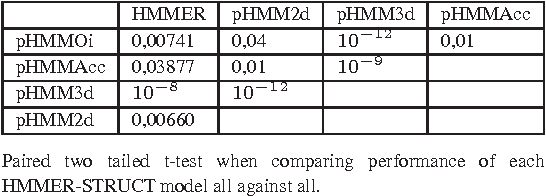
Motivation: Profile hidden Markov Models (pHMMs) are a popular and very useful tool in the detection of the remote homologue protein families. Unfortunately, their performance is not always satisfactory when proteins are in the 'twilight zone'. We present HMMER-STRUCT, a model construction algorithm and tool that tries to improve pHMM performance by using structural information while training pHMMs. As a first step, HMMER-STRUCT constructs a set of pHMMs. Each pHMM is constructed by weighting each residue in an aligned protein according to a specific structural property of the residue. Properties used were primary, secondary and tertiary structures, accessibility and packing. HMMER-STRUCT then prioritizes the results by voting. Results: We used the SCOP database to perform our experiments. Throughout, we apply leave-one-family-out cross-validation over protein superfamilies. First, we used the MAMMOTH-mult structural aligner to align the training set proteins. Then, we performed two sets of experiments. In a first experiment, we compared structure weighted models against standard pHMMs and against each other. In a second experiment, we compared the voting model against individual pHMMs. We compare method performance through ROC curves and through Precision/Recall curves, and assess significance through the paired two tailed t-test. Our results show significant performance improvements of all structurally weighted models over default HMMER, and a significant improvement in sensitivity of the combined models over both the original model and the structurally weighted models.