 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Multiple Filter Based Neural Network Approach to the Extrapolation of Adsorption Energies on Metal Surfaces for Catalysis Applications
Paper and Code
Oct 01, 2019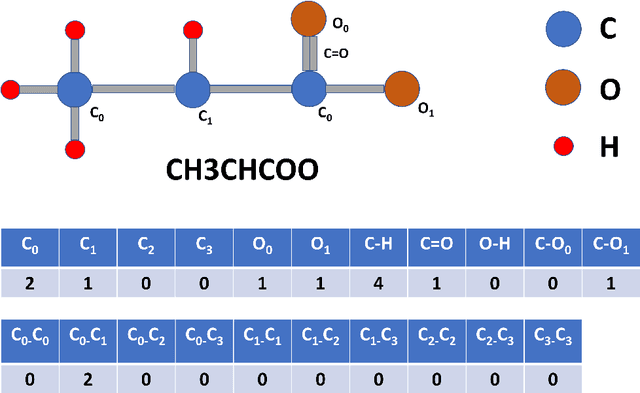
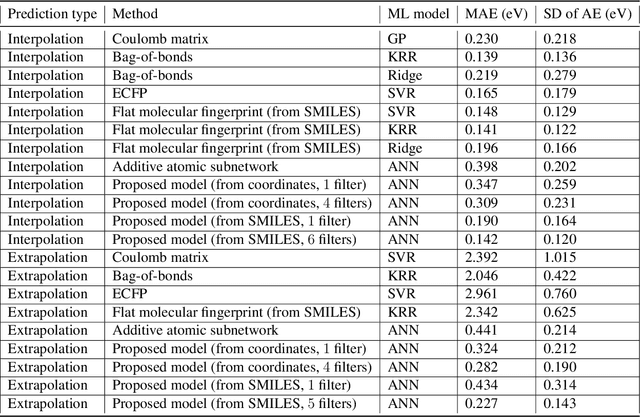
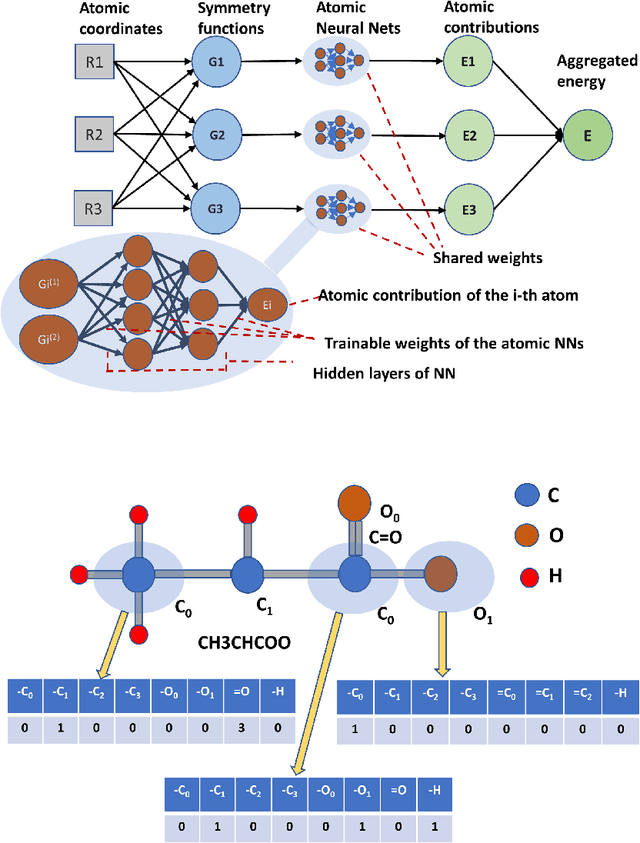
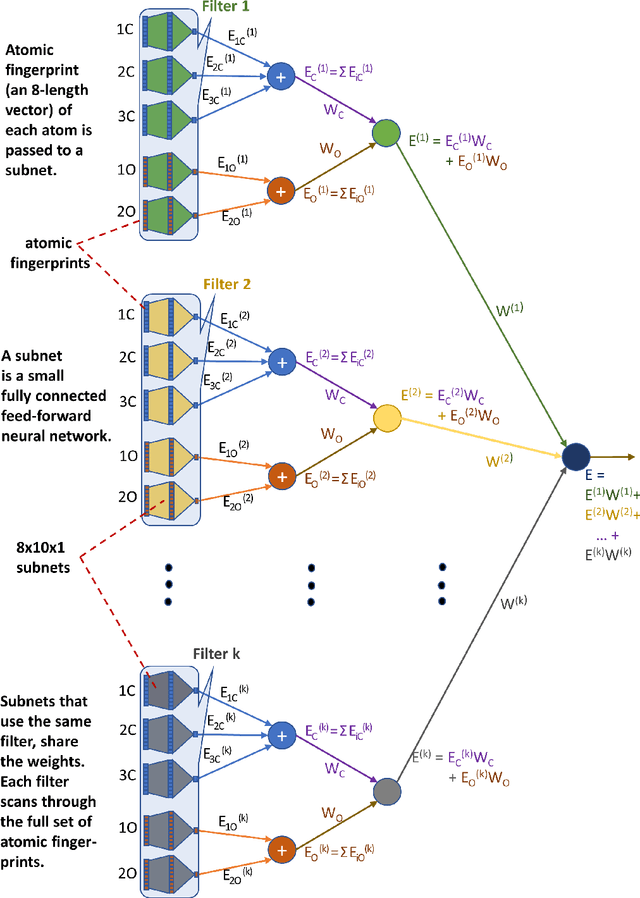
Computational catalyst discovery involves the development of microkinetic reactor models based on estimated parameters determined from density functional theory (DFT). For complex surface chemistries, the cost of calculating the adsorption energies by DFT for a large number of reaction intermediates can become prohibitive. Here, we have identified appropriate descriptors and machine learning models that can be used to predict part of these adsorption energies given data on the rest of them. Our investigations also included the case when the species data used to train the predictive model is of different size relative to the species the model tries to predict - an extrapolation in the data space which is typically difficult with regular machine learning models. We have developed a neural network based predictive model that combines an established model with the concepts of a convolutional neural network that, when extrapolating, achieves significant improvement over the previous models.