 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine-Learning Prediction of the Computed Band Gaps of Double Perovskite Materials
Jan 04, 2023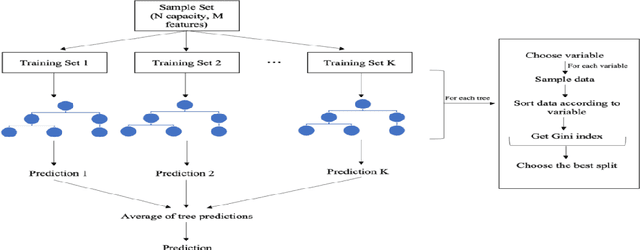
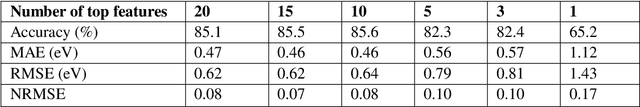
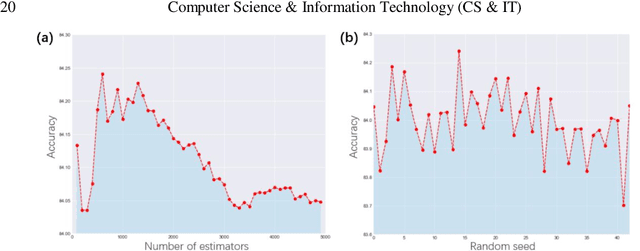
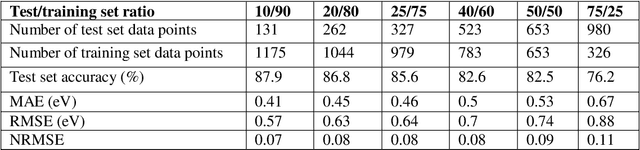
Prediction of the electronic structure of functional materials is essential for the engineering of new devices. Conventional electronic structure prediction methods based on density functional theory (DFT) suffer from not only high computational cost, but also limited accuracy arising from the approximations of the exchange-correlation functional. Surrogate methods based on machine learning have garnered much attention as a viable alternative to bypass these limitations, especially in the prediction of solid-state band gaps, which motivated this research study. Herein, we construct a random forest regression model for band gaps of double perovskite materials, using a dataset of 1306 band gaps computed with the GLLBSC (Gritsenko, van Leeuwen, van Lenthe, and Baerends solid correlation) functional. Among the 20 physical features employed, we find that the bulk modulus, superconductivity temperature, and cation electronegativity exhibit the highest importance scores, consistent with the physics of the underlying electronic structure. Using the top 10 features, a model accuracy of 85.6% with a root mean square error of 0.64 eV is obtained, comparable to previous studies. Our results are significant in the sense that they attest to the potential of machine learning regressions for the rapid screening of promising candidate functional materials.