 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine learning driven search of hydrogen storage materials
Mar 06, 2025
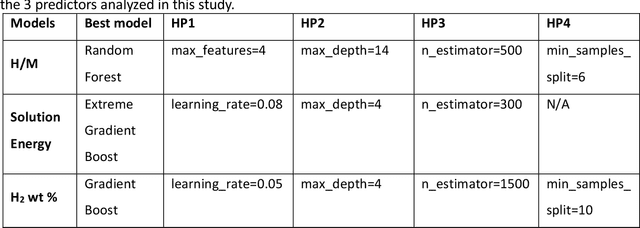
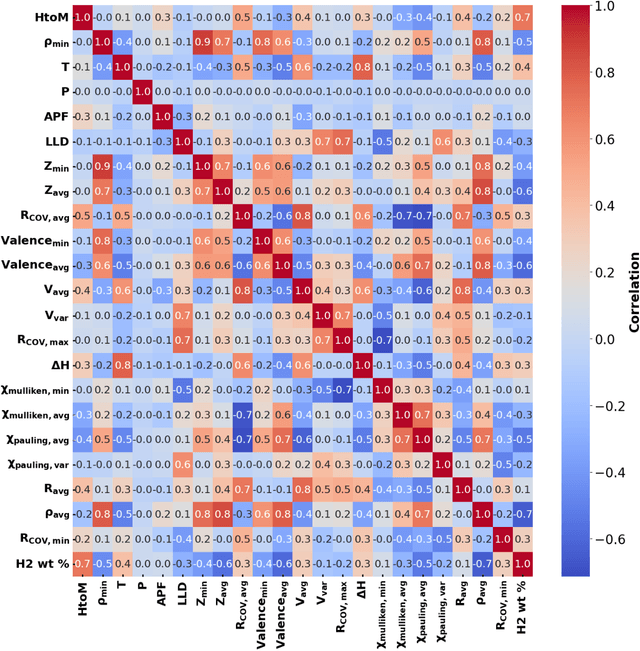

The transition to a low-carbon economy demands efficient and sustainable energy-storage solutions, with hydrogen emerging as a promising clean-energy carrier and with metal hydrides recognized for their hydrogen-storage capacity. Here, we leverage machine learning (ML) to predict hydrogen-to-metal (H/M) ratios and solution energy by incorporating thermodynamic parameters and local lattice distortion (LLD) as key features. Our best-performing ML model provides improvements to H/M ratios and solution energies over a broad class of ternary alloys (easily extendable to multi-principal-element alloys), such as Ti-Nb-X (X = Mo, Cr, Hf, Ta, V, Zr) and Co-Ni-X (X = Al, Mg, V). Ti-Nb-Mo alloys reveal compositional effects in H-storage behavior, in particular Ti, Nb, and V enhance H-storage capacity, while Mo reduces H/M and hydrogen weight percent by 40-50%. We attributed to slow hydrogen kinetics in molybdenum rich alloys, which is validated by our pressure-composition isotherm (PCT) experiments on pure Ti and Ti5Mo95 alloys. Density functional theory (DFT) and molecular simulations also confirm that Ti and Nb promote H diffusion, whereas Mo hinders it, highlighting the interplay between electronic structure, lattice distortions, and hydrogen uptake. Notably, our Gradient Boosting Regression model identifies LLD as a critical factor in H/M predictions. To aid material selection, we present two periodic tables illustrating elemental effects on (a) H2 wt% and (b) solution energy, derived from ML, and provide a reference for identifying alloying elements that enhance hydrogen solubility and storage.