 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePerspective: Towards sustainable exploration of chemical spaces with machine learning
Mar 31, 2026Artificial intelligence is transforming molecular and materials science, but its growing computational and data demands raise critical sustainability challenges. In this Perspective, we examine resource considerations across the AI-driven discovery pipeline--from quantum-mechanical (QM) data generation and model training to automated, self-driving research workflows--building on discussions from the ``SusML workshop: Towards sustainable exploration of chemical spaces with machine learning'' held in Dresden, Germany. In this context, the availability of large quantum datasets has enabled rigorous benchmarking and rapid methodological progress, while also incurring substantial energy and infrastructure costs. We highlight emerging strategies to enhance efficiency, including general-purpose machine learning (ML) models, multi-fidelity approaches, model distillation, and active learning. Moreover, incorporating physics-based constraints within hierarchical workflows, where fast ML surrogates are applied broadly and high-accuracy QM methods are used selectively, can further optimize resource use without compromising reliability. Equally important is bridging the gap between idealized computational predictions and real-world conditions by accounting for synthesizability and multi-objective design criteria, which is essential for practical impact. Finally, we argue that sustainable progress will rely on open data and models, reusable workflows, and domain-specific AI systems that maximize scientific value per unit of computation, enabling efficient and responsible discovery of technological materials and therapeutics.
Graph-neural-network predictions of solid-state NMR parameters from spherical tensor decomposition
Dec 19, 2024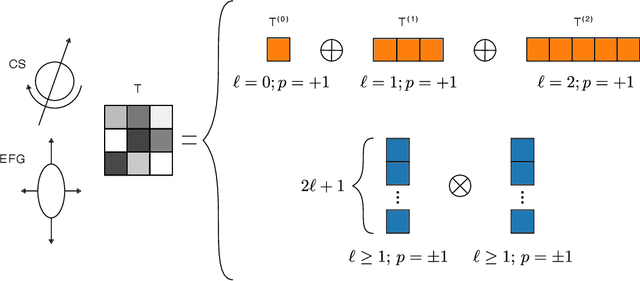
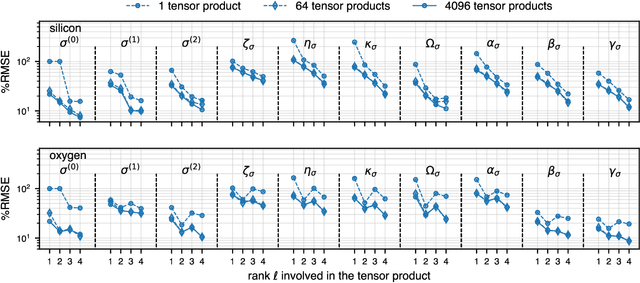
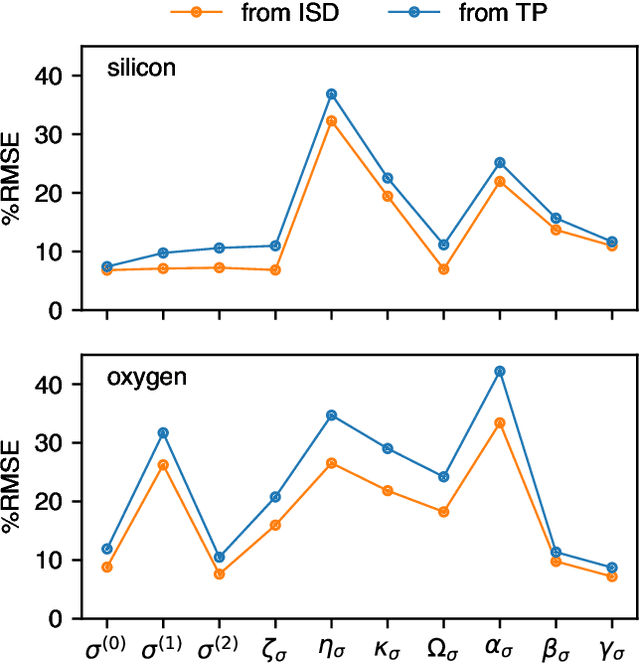
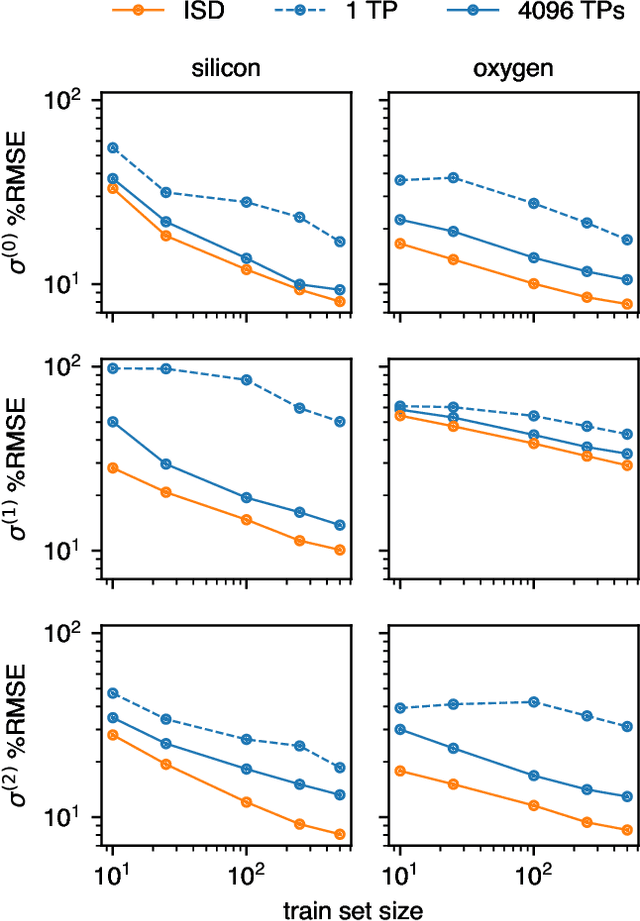
Nuclear magnetic resonance (NMR) is a powerful spectroscopic technique that is sensitive to the local atomic structure of matter. Computational predictions of NMR parameters can help to interpret experimental data and validate structural models, and machine learning (ML) has emerged as an efficient route to making such predictions. Here, we systematically study graph-neural-network approaches to representing and learning tensor quantities for solid-state NMR -- specifically, the anisotropic magnetic shielding and the electric field gradient. We assess how the numerical accuracy of different ML models translates into prediction quality for experimentally relevant NMR properties: chemical shifts, quadrupolar coupling constants, tensor orientations, and even static 1D spectra. We apply these ML models to a structurally diverse dataset of amorphous SiO$_2$ configurations, spanning a wide range of density and local order, to larger configurations beyond the reach of traditional first-principles methods, and to the dynamics of the $\alpha\unicode{x2013}\beta$ inversion in cristobalite. Our work marks a step toward streamlining ML-driven NMR predictions for both static and dynamic behavior of complex materials, and toward bridging the gap between first-principles modeling and real-world experimental data.
Synthetic pre-training for neural-network interatomic potentials
Jul 24, 2023Machine learning (ML) based interatomic potentials have transformed the field of atomistic materials modelling. However, ML potentials depend critically on the quality and quantity of quantum-mechanical reference data with which they are trained, and therefore developing datasets and training pipelines is becoming an increasingly central challenge. Leveraging the idea of "synthetic" (artificial) data that is common in other areas of ML research, we here show that synthetic atomistic data, themselves obtained at scale with an existing ML potential, constitute a useful pre-training task for neural-network interatomic potential models. Once pre-trained with a large synthetic dataset, these models can be fine-tuned on a much smaller, quantum-mechanical one, improving numerical accuracy and stability in computational practice. We demonstrate feasibility for a series of equivariant graph-neural-network potentials for carbon, and we carry out initial experiments to test the limits of the approach.
Synthetic data enable experiments in atomistic machine learning
Nov 29, 2022Machine-learning models are increasingly used to predict properties of atoms in chemical systems. There have been major advances in developing descriptors and regression frameworks for this task, typically starting from (relatively) small sets of quantum-mechanical reference data. Larger datasets of this kind are becoming available, but remain expensive to generate. Here we demonstrate the use of a large dataset that we have "synthetically" labelled with per-atom energies from an existing ML potential model. The cheapness of this process, compared to the quantum-mechanical ground truth, allows us to generate millions of datapoints, in turn enabling rapid experimentation with atomistic ML models from the small- to the large-data regime. This approach allows us here to compare regression frameworks in depth, and to explore visualisation based on learned representations. We also show that learning synthetic data labels can be a useful pre-training task for subsequent fine-tuning on small datasets. In the future, we expect that our open-sourced dataset, and similar ones, will be useful in rapidly exploring deep-learning models in the limit of abundant chemical data.