 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDirectMultiStep: Direct Route Generation for Multi-Step Retrosynthesis
May 22, 2024



Traditional computer-aided synthesis planning (CASP) methods rely on iterative single-step predictions, leading to exponential search space growth that limits efficiency and scalability. We introduce a transformer-based model that directly generates multi-step synthetic routes as a single string by conditionally predicting each molecule based on all preceding ones. The model accommodates specific conditions such as the desired number of steps and starting materials, outperforming state-of-the-art methods on the PaRoutes dataset with a 2.2x improvement in Top-1 accuracy on the n$_1$ test set and a 3.3x improvement on the n$_5$ test set. It also successfully predicts routes for FDA-approved drugs not included in the training data, showcasing its generalization capabilities. While the current suboptimal diversity of the training set may impact performance on less common reaction types, our approach presents a promising direction towards fully automated retrosynthetic planning.
Kernel-Elastic Autoencoder for Molecular Design
Oct 12, 2023


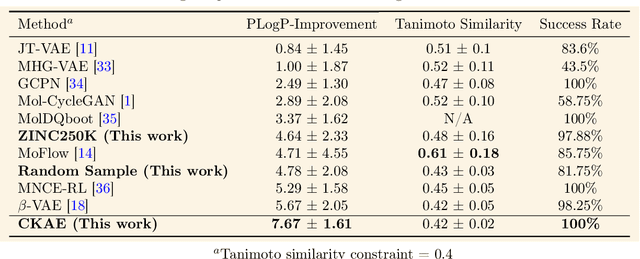
We introduce the Kernel-Elastic Autoencoder (KAE), a self-supervised generative model based on the transformer architecture with enhanced performance for molecular design. KAE is formulated based on two novel loss functions: modified maximum mean discrepancy and weighted reconstruction. KAE addresses the long-standing challenge of achieving valid generation and accurate reconstruction at the same time. KAE achieves remarkable diversity in molecule generation while maintaining near-perfect reconstructions on the independent testing dataset, surpassing previous molecule-generating models. KAE enables conditional generation and allows for decoding based on beam search resulting in state-of-the-art performance in constrained optimizations. Furthermore, KAE can generate molecules conditional to favorable binding affinities in docking applications as confirmed by AutoDock Vina and Glide scores, outperforming all existing candidates from the training dataset. Beyond molecular design, we anticipate KAE could be applied to solve problems by generation in a wide range of applications.
Using Restricted Boltzmann Machines to Model Molecular Geometries
Dec 13, 2020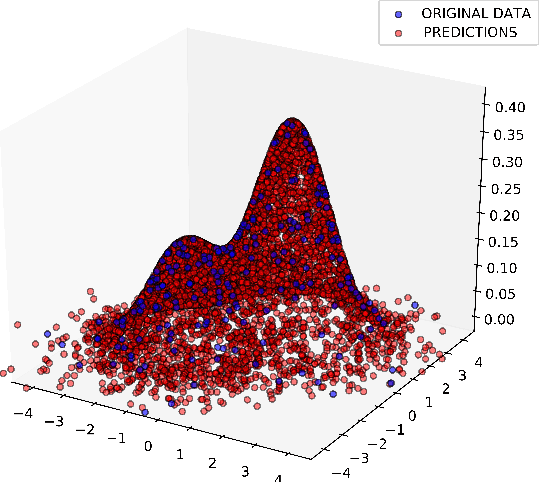

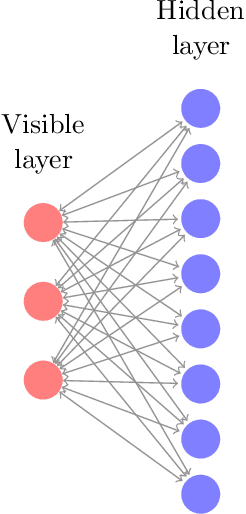
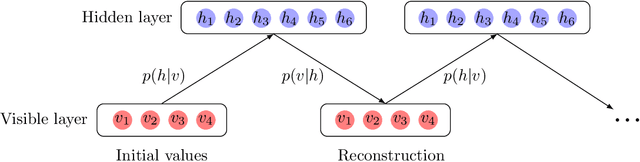
Precise physical descriptions of molecules can be obtained by solving the Schrodinger equation; however, these calculations are intractable and even approximations can be cumbersome. Force fields, which estimate interatomic potentials based on empirical data, are also time-consuming. This paper proposes a new methodology for modeling a set of physical parameters by taking advantage of the restricted Boltzmann machine's fast learning capacity and representational power. By training the machine on ab initio data, we can predict new data in the distribution of molecular configurations matching the ab initio distribution. In this paper we introduce a new RBM based on the Tanh activation function, and conduct a comparison of RBMs with different activation functions, including sigmoid, Gaussian, and (Leaky) ReLU. Finally we demonstrate the ability of Gaussian RBMs to model small molecules such as water and ethane.