 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Fair Experimental Comparison of Neural Network Architectures for Latent Representations of Multi-Omics for Drug Response Prediction
Aug 31, 2022
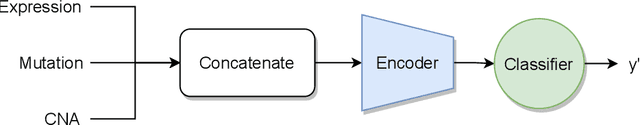

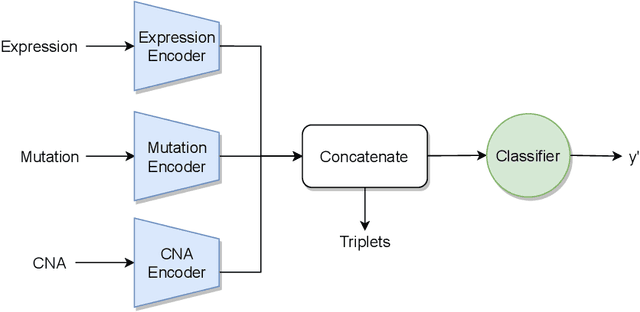
Recent years have seen a surge of novel neural network architectures for the integration of multi-omics data for prediction. Most of the architectures include either encoders alone or encoders and decoders, i.e., autoencoders of various sorts, to transform multi-omics data into latent representations. One important parameter is the depth of integration: the point at which the latent representations are computed or merged, which can be either early, intermediate, or late. The literature on integration methods is growing steadily, however, close to nothing is known about the relative performance of these methods under fair experimental conditions and under consideration of different use cases. We developed a comparison framework that trains and optimizes multi-omics integration methods under equal conditions. We incorporated early integration and four recently published deep learning methods: MOLI, Super.FELT, OmiEmbed, and MOMA. Further, we devised a novel method, Omics Stacking, that combines the advantages of intermediate and late integration. Experiments were conducted on a public drug response data set with multiple omics data (somatic point mutations, somatic copy number profiles and gene expression profiles) that was obtained from cell lines, patient-derived xenografts, and patient samples. Our experiments confirmed that early integration has the lowest predictive performance. Overall, architectures that integrate triplet loss achieved the best results. Statistical differences can, overall, rarely be observed, however, in terms of the average ranks of methods, Super.FELT is consistently performing best in a cross-validation setting and Omics Stacking best in an external test set setting. The source code of all experiments is available under \url{https://github.com/kramerlab/Multi-Omics_analysis}