 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSPT-NRTL: A physics-guided machine learning model to predict thermodynamically consistent activity coefficients
Sep 09, 2022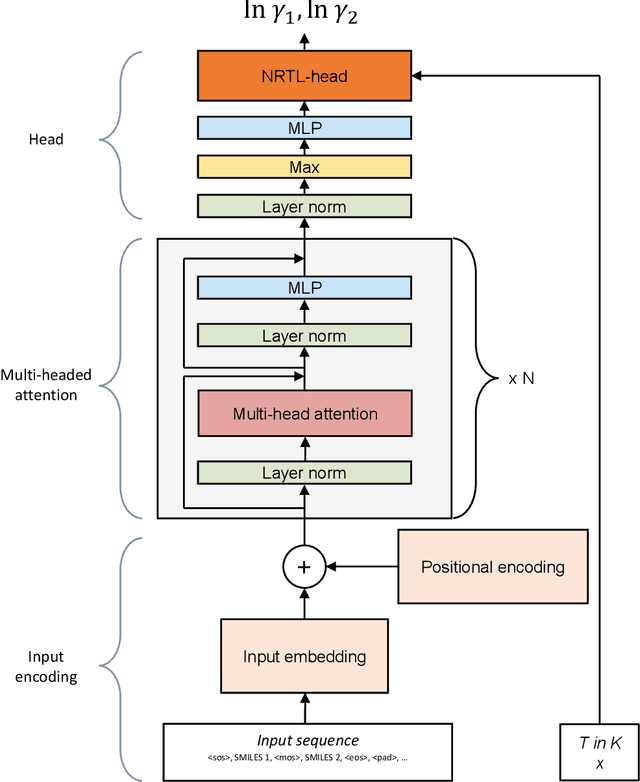
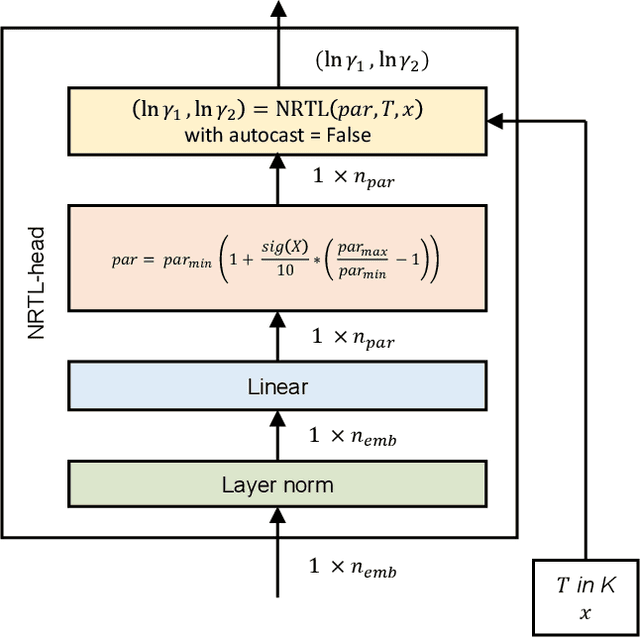


The availability of property data is one of the major bottlenecks in the development of chemical processes, often requiring time-consuming and expensive experiments or limiting the design space to a small number of known molecules. This bottleneck has been the motivation behind the continuing development of predictive property models. For the property prediction of novel molecules, group contribution methods have been groundbreaking. In recent times, machine learning has joined the more established property prediction models. However, even with recent successes, the integration of physical constraints into machine learning models remains challenging. Physical constraints are vital to many thermodynamic properties, such as the Gibbs-Dunham relation, introducing an additional layer of complexity into the prediction. Here, we introduce SPT-NRTL, a machine learning model to predict thermodynamically consistent activity coefficients and provide NRTL parameters for easy use in process simulations. The results show that SPT-NRTL achieves higher accuracy than UNIFAC in the prediction of activity coefficients across all functional groups and is able to predict many vapor-liquid-equilibria with near experimental accuracy, as illustrated for the exemplary mixtures water/ethanol and chloroform/n-hexane. To ease the application of SPT-NRTL, NRTL-parameters of 100 000 000 mixtures are calculated with SPT-NRTL and provided online.